Chronic Myelomonocytic Leukemia; Rare Blast Transformation: Case Report with Literature Review
Author'(s): Giamal Edin Gmati1,4*, Lubna AlZadjali2, Nahlah AlGhasham2, Areej Almugairi2, Ahmad Alzghoul2, Yazeed Althobaiti3, Azizah Alkhaldi3 and Mohammed Albalwi3,4
1Adult Hematology & HSCT Division, Department of Oncology,King Abdulaziz Medical City, Riyadh. KSA.
2Hematology division, Department of Pathology and Laboratory Medicine, King Abdulaziz Medical City, Riyadh. KSA.
3Molecular division, Department of Pathology and Laboratory Medicine, King Abdulaziz Medical City, Riyadh. KSA.
4King Saud Bin Abdulaziz University for Health Sciences,Riyadh. KSA.
*Correspondence:
G E Gmati, Adult Hematology & HSCT Division, Mail code 1775, Department of Oncology, King Abdulaziz Medical City,Riyadh 11426. KSA.
Received: 15 October 2020; Accepted: 01 November 2020
Citation: Gmati GE, AlZadjali L, AlGhasham N, et al. Chronic Myelomonocytic Leukemia; Rare Blast Transformation: Case Report with Literature Review. Cancer Sci Res. 2020; 3(1): 1-7.
Abstract
Chronic myelomonocytic leukemia (CMML) is a malignant hematopoietic stem cell disorder with clinical and pathological features of both a myeloproliferative neoplasm (MPN) and myelodysplastic syndrome (MDS). CMML is characterized by peripheral blood monocytosis accompanied by bone marrow dysplasia; cytopenias and hepatosplenomegaly are common. The pathogenesis of CMML is poorly understood. CMML arises by the serial acquisition of somatic genetic events that create multiple, distinct neoplastic cell clones. Transformation to acute myeloid leukemia (AML) is a well-known phenomenon and reaches 15-20% of cases. However, we report here a rare type of transformation to B cell acute lymphoblastic leukemia (B-ALL) in a 71-year-old female. There is a paucity of case reports on this particular transformation where only one case in adults and another case in a 10-month-old infant. The transformation of other subtypes of MDS such as refractory anemia with or without sideroblasts and or with increased blasts to B-ALL has been reported in the past. It is not very clear if there is a specific provocative factor for this particular transformation since the cytogenetic and molecular markers in our patients were all negative except pathogenic variants in the NRAS and TET2 genes at high allele frequencies on retrospective analysis. The new lymphoid malignancy /clone for our case was cleared after 2 cycles of mini HCVAD protocol. We focused our treatment on this new lymphoid clone which was cleared after 2 cycles of mini HCVAD protocol. Although the transformation of CMML into B-ALL is a rare complication, we believe further analysis or cohort study on larger CMML cases to elaborate more on the risk factors, cytogenetic and molecular events that might contribute to such complication.
Keywords
Introduction
Chronic myelomonocytic leukemia (CMML) is a clonal hematopoietic stem cell-derived disorder characterized by abnormal bone marrow (BM) production of monocytes resulting in absolute (≥1×10 9/L) and relative (>10%) peripheral blood (PB) monocytosis, dysplasia in any or all hematopoietic cell lineages [1].
CMML is the most frequent entity among myeloproliferative/ myelodysplastic syndromes, as defined by the World Health Organization (WHO) classification of myeloid malignancies in 2008 [2]. CMML was recognized as a distinct entity by the French-American-British (FAB) group in 1976 [3]. Based on the percentage of blast cells in the bone marrow and peripheral blood, CMML is further stratified into CMML-1 (< 5% in blood, < 10% in the bone marrow) and CMML-2 (5 to 19% in the blood; 10 to 19% in the bone marrow, or less if Auer inclusions are present) [4].
The median age at diagnosis of CMML is 65 to 75 years with moderate male predominance. The actual incidence of CMML may be higher than that predicted by cancer databases since the nonspecific symptoms may evade detection in the early stages of the disease, while other cases may not undergo definitive testing, such as bone marrow biopsy, due to comorbidities and very advanced age [5]. Clinical presentation of CMML patients can be non-specific and the disease would be suspected upon the initial blood work up when changes of anemia, neutropenia, monocytosis and thrombocytopenia are detected. However, splenomegaly tends to occur in up to 25% of patients and is often accompanied by hepatomegaly, lymphadenopathy, or nodular cutaneous leukemic infiltrates [6,7].
Case history
Seventy-one year old Saudi female from the northern region presented in March 2018 with bleeding per gum and skin ecchymosis. She is known to have type 2 diabetes mellitus and hypothyroidism.
Clinical examination revealed scattered skin ecchymosis and petechial spots mostly on upper limbs. Hepatosplenomegaly was evident with enlarged spleen 10 cm below left costal margin and, liver of 15 cm below right costal margin.
Complete blood count (CBC) on admission showed Hb of 70 g/l, MCV 87fl, WBC 5.1 x10^9/L, ANC 1.9 x10^9/L, lymphocytes1.6 x10^9/L, monocytes 1.3 x10^9/L (25 %) and Platelets 39 x10^9/L. Peripheral Blood smear showed left shifted granulocytes, occasional Pseudo Pelger-Huet neutrophils, rare medium- sized circulating blasts with high nuclear/cytoplasmic (N/C) ratio and some monocytes. Few large platelets were noted with no platelet aggregates (Figure 1a).
Bone marrow was performed and revealed hyper cellular marrow for patient`s age with around 80% cellularity, adequate trilineage hematopoiesis with dysplasia mainly noted in erythroid precursors in the form of significant megaloblastoid changes and basophilic stippling. Some small and hypolobated megakaryocytes (small and hypolobated) precursors. The blasts are increased and account for around 12% of marrow nucleated cells, while the monocytes account for 1.2% (Figure 1b).
The Flow cytometry analysis from bone marrow detected around 1% myeloblast expressing positivity for CD34, CD117, CD13, CD33, and partial positivity for CD7. Interestingly, there were about 3.6% B lymphoblasts with a hematogone pattern of expression for CD19, CD10, CD20, CD79a, CD22 and TdT.
Conventional cytogenetic showed normal female karyotype (46; XX) and extensive FISH panel for myelodysplasia and myeloproliferative neoplasm disorder including BCR/ABL1, PDGFRA and PDGFRB were performed and did not demonstrate positive abnormalities. Likewise molecular analysis for NPM1, FLT3-ITD and KIT mutations were all negative. However, molecular testing for myeloid tumor panel was retrospectively requested. A presence of pathogenic variants in the NRAS and TET2 genes at high allele frequencies are detected.
In view of the patient`s presentation of cytopenia, splenomegaly, persistent peripheral blood monocytosis (>10% of total WBC) with no clinical evidence of infection or other malignancy, significant bone marrow dysplasia in two cell lineages , negative mutations for BCR-ABL1, JAK2 and NPM1 and absence of rearrangement of PDGFRA, PDGFRB, FGR1, the diagnosis of Myelodysplastic / Myeloproliferative neoplasm in the form of chronic myelomonocytic leukemia (CMML) was made. Given the percentage of bone marrow blasts less than 20 % the patient was diagnosed with chronic myelomonocytic leukemia-2 (CMML-2).
The patient was started on Azacitidine 100 mg s/c daily for one week/month in April 2018 for a total of 13 months. Despite the fact that her count did not improve dramatically, the patient herself felt more energetic and active. In addition, most of the skin ecchymosis and bruises have disappeared and no more spontaneous bleeding episodes.
The patient requested treatment break after cycle 13, she remained mildly pancytopenic with persistent hepatosplenomegaly.
A follow up marrow biopsy in January 2019 showed increased cellularity to 90% with no immunomorphological evidence for disease progression as blasts were 10-11% and the picture as a whole still consistent with CMML-2. Flow cytometry, Cytogenetic and FISH analysis showed no new changes compared to the bone marrow at presentation one year earlier.
Since there was no evidence of disease progression and given the fact that the patient was enjoying a reasonable quality of life, she decided to stay off treatment with regular clinic visits and assessment.
Due to worsened anemia and thrombocytopenia, a bone marrow was performed in February 2020 that showed raised blast count to 46%. These blasts unexpectedly resemble lymphoblast by morphology (Figure 2a and b). The flow cytometry detected around 20% blasts expressing CD 34, CD22, HLA-DR, CD10, CD19, cytoCD79a and TdT, the immunophenotypic profile of B lymphoblastic leukemia (B- ALL). The hyper cellular bone marrow trephine revealed a large collection of small to medium size blasts of which the immunohistochemistry also concord with flow cytometry finding for the presence of lymphoblast (Figures 2c and d).
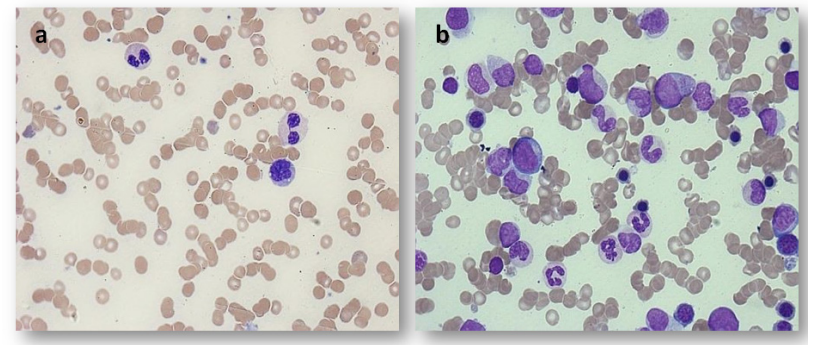
Figure 1: Peripheral blood smear and bone marrow in March 2018 consistent with a diagnosis of chronic myelomonocytic leukemia. a: Patient peripheral blood smear showing monocyte and two Pseudo Pelger-Huet neutrophils. b: Patient bone marrow aspirate showing hypercellularity with prominent monocytes precursors, dysplastic granulocyte precursors and increased blasts (× 50 high power magnification).
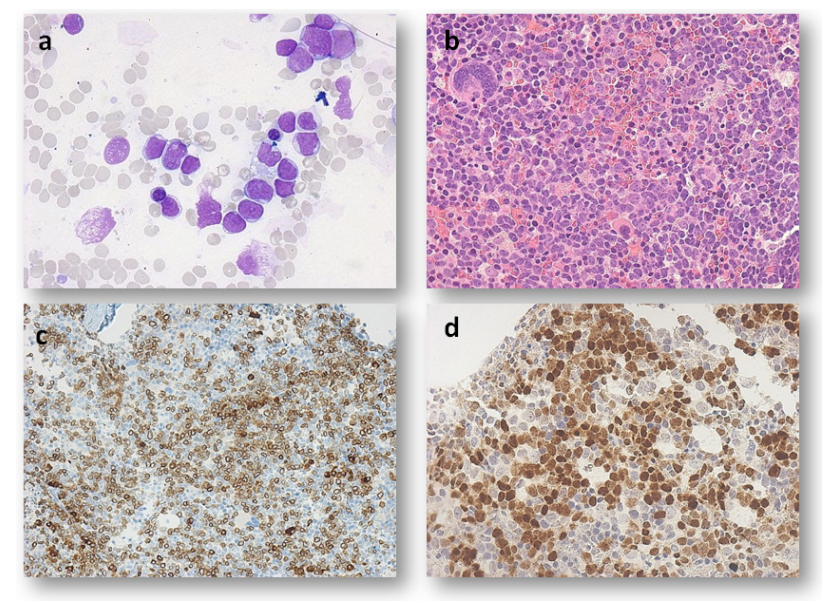
Figure 2: Microscopic examination of the bone marrow in February 2020. a: Bone marrow aspirates showing medium size lymphoblast with scanty cytoplasm (× 50 high power magnifications). b: Bone marrow biopsy showing large collection of small to medium size blasts (× 40 high power magnification). c: Blasts with positivity for CD79a immunohistochemical stain (× 20 high power magnification). d: Blasts with positivity for TdT immunohistochemical stain (× 40 high power magnification).
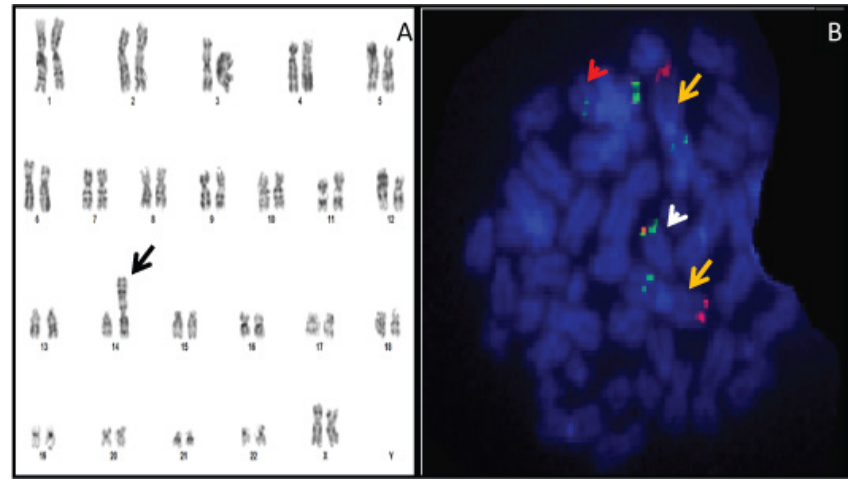
Figure 3: FISH and Cytogenetic Karyotyping images from patient Bone Marrow culture in 2020. A) Cytogenetic karyotyping revealed a female with 46, XX, der (14) t (1; 14) (q21; p11.1) (Black arrow). B) A metaphase hybridized with combined probes for 1p36 (Red)/1q25 (Green) and IGH gene (Dual color of Green/Red) on chromosome 14. Yellow arrows indicate presence of two normal chromosome1 while the white arrow indicate presence of one normal chromosome 14 and the red arrow indicate the derivation chromosome of 14 involved combining of extra segment of the long arm chromosome 1 with the other homologue of chromosome 14.
Bone marrow Chromosome analysis revealed female karyotype with unbalanced translocation between long arms of chromosome 1 and 14 in 5% of all examined cells Karyotype: 46,XX,der(14) t(1;14)(q21;p11.1)[2]/46,XX[18]. This finding was also confirmed by a FISH study that revealed an extra copy of 1q25 region in 2.4 % of examined cells, based on metaphases third copy of 1q25 region showed on chromosome 14 (Figure 3A and B).
The FISH for B lymphoblastic and myeloid leukemia panel did not detect any positive abnormalities. With the new developments of our patient disease pathology, the management plan had to be changed to target the new leukemia clone of B-ALL where she received two rounds of mini HCVAD protocol. Disease progression evaluation by bone marrow examination revealed resolution of the B-ALL clone and the return to CMML 2 immunomorphological status.
Discussion
CMML is a relatively rare disease, with an approximate incidence of 0.3 to 0.7per 100,000-person-year, median age at diagnosis of 71–74 years, and a strong male predominance (M: F ratio, 1.5/3:1) [8-11]. Clinical heterogeneity is broadly captured by the historical categorization into proliferative type (MPN-CMML) and dysplastic type (MDS-CMML) CMML, based on the presence of a white blood cell (WBC) count ≥13×109/L in the former [12].
The 2016 World Health Organization (WHO) criteria for the diagnosis of CMML include all of the following: 1) Persistent (≥3 months) peripheral blood monocytosis; absolute monocyte count >1000/microlitre and greater than 10 percent of the entire white blood cell differential. 2) Not meeting WHO criteria for BCR- ABL1 positive chronic myeloid leukemia, primary myelofibrosis, polycythemia vera, or essential thrombocytosis. 3) <20 percent myeloblasts, monoblasts and promonocytes in peripheral blood and bone marrow. 4) Dysplastic changes in one or more myeloid lineages. If myelodysplastic changes are absent or minimal, the diagnosis of CMML can be made if the above three criteria are met and: a) An acquired clonal cytogenetic (or mutational abnormality) is present in bone marrow cells or b) Persistent monocytosis for ≥3 months and all other causes of monocytosis have been excluded [12]. The WHO classification sub classifies cases into one of three groups based on prognostic value: 1) CMML 0, <2 percent blasts in the peripheral blood and <5 percent blasts in the marrow 2) CMML1, 2 to 4 percent blasts in the peripheral blood and/or 5 to 9 percent blasts in the marrow 3) CMML 2, 5 to 19 percent blasts in the peripheral blood, 10 to 19 percent blasts in the marrow, or presence of one or more Auer rods [14]. Cytogenetic abnormalities occur in approximately 30% of patients with CMML. Common abnormalities include trisomy 8, -Y, abnormalities of chromosome 7 (monosomy 7 and or del 7q), trisomy 21 and complex karyotype. Cytogenetic risk stratification by the Spanish group was developed and categorized patients into three groups: high risk (trisomy 8, chromosome 7 abnormalities and complex karyotype), intermediate-risk (all chromosomal abnormalities except those in high and low risk groups) and low risk (all normal karyotype or –Y) with 5 year overall survival (OS) of 4%, 26% and 35 % respectively [15]. Trisomy 1q is known to play part on a complex chromosome change affecting the subtypes of MDS other than CMML. Rare reports detect this abnormality in CMML [16]. Gene mutations occur in approximately 90% of patients with CMML [17], and can be divided according to the major class of these mutations (Table 1).

The most frequent mutations in patients with CMML are TET2, SRSF2, ASXL1 and RAS pathway with a frequency of 60%, 50%, 40% and 30% respectively [17,31]. Leukemic transformation of CMML to AML is associated with risk factors such as high-risk karyotype, PB blast percentage, absolute monocytic count (AMC)>10 × 109/L, ASXL1, RUNX1, NRAS, SETBP1, DNMT3A and NPM1 mutations [32-34]. This transformation is usually associated with poor outcome, 1, 3,5 year survival rates of 25%, 9% and 6% respectively with OS of 6 months in one large study [35]. Blast transformation to lymphoid malignancies is generally rare; however, few cases of transformation into monoclonal gammopathy of uncertain significance, multiple myeloma, chronic lymphocytic leukemia and angioimmunoblastic lymphoma have been reported [36-40]. Although the transformation of CMML to acute lymphoblastic leukemia is rare, several cases of MDS transformed into B-ALL in adults have been reported [41]. Infantile CMML suggested being clonal disorders arising in a pluripotent stem cell that can also differentiate along the lymphoid cell lineage [42].
In our patient high allele frequencies of NRAS and TET2 genes were detected. TET2 mutations can be detected in both myeloid and lymphoid malignancies [43]. In TET2-mutated lymphoid malignancies, the detection of a mutated allele in immature myeloid progenitors suggest that a common genetic background could generate both myeloid and lymphoid diseases [44,45]. The presence of new cytogenetic abnormality of t(1;14) later at diagnosis of B-ALL and on subsequent bone marrow after disappearance of lymphoblast clone and its correlation with the blast transformation is not yet clearly understood. The use of azacitidine in the treatment of CMML was approved by FDA after early trials have shown activity in the treatment of myelodysplastic syndromes where CMML represented a minority of the study population [46]. Due to the fact that lymphoid malignancy transformation in CMML patients is rare, no specific guidelines on how these transformations should be managed. Nevertheless, on implementing the standard of care in the management of B-ALL, our patient went into remission after receiving a modified HCVAD protocol which is the usual scenario given the fact that it was not associated with recurrent cytogenetic or molecular markers.
Conclusion
CMML is a bone marrow disorder characterized by myeloproliferative and myelodysplastic features. Comprehensive mutational studies based mainly on next-generation sequencing technologies have scripted the mutational landscape of CMML and identified recurrently mutated genes involved in epigenetic regulation, pre-messenger RNA splicing, and cell signaling. Leukemic transformation usually to AML is a well-known complication. Lymphblastic transformation is a rare complication with a handful of reports in the literature. Further investigations and deep analysis warranted on this particular transformation.
References
- Coltro G, Patnaik MM. Chronic Myelomonocytic Leukemia Insights into Biology Prognostic Factors and Current Oncology Reports. 2019; 21: 101.
- Vardiman JW, Thiele J, Arber DA, et The 2008 revision of the World Health Organisation WHO classification of myeloid neoplasms and acute leukemia rational and important changes. Blood. 2009; 114: 937-951.
- Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the acute French-American-British FAB co-operative group. Br J Haematol. 1976; 33: 451.
- Vardiman JW, Harris NL, Brunning RD. The World Health Organization WHO Classification of the Myeloid Blood. 2002; 100: 2292-2302.
- McQuilten ZK, Wood EM, Polizzotto MN, et Underestimation of Myelodysplastic Syndrome Incidence by Cancer Registries: Results from a Population-Based Data Linkage Study Cancer. 2014; 120: 1686-1694.
- Geary CG, Catovsky D, Wiltshaw E, et ChronicMyelomonocytic Leukemia. Br J Haematol. 1975; 30: 289.
- Duguid JK, Mackie MJ, McVerry Skin Infiltration Associated with Chronic Myelomonocytic Leukemia. Br J Haematol. 1983; 53: 257-264.
- Rollison DE, Howlader N, Smith MT, et al. Epidemiology of myelodysplastic syndromes and chronic myeloproliferative disorders in the United States 20012004 using data from the NAACCR and SEER Blood. 2008; 112: 45-52.
- Benzarti S, Daskalakis M, Feller A, et al. NICER Working Trends of incidence and survival of patients with chronic myelomonocytic leukemia between 1999 and 2014 a comparison between Swiss and American population-based cancer registries. Cancer Epidemiol. 2019; 59: 51-57.
- Such E, Germing U, Malcovati L, et al. Development and validation of a prognostic scoring system for patients with chronic myelomonocytic leukemia. Transfuze a Hematologie 2013; 19: 191.
- Patnaik MM, Lasho TL, Finke CM, et Spliceosome mutations involving SRSF2 SF3B1 andU2AF35inchronicmyelomonocytic leukemia prevalence clinical correlates and prognostic relevance. Am J Hematol. 2013; 88: 201-206.
- Bennett JM, Catovsky D, Daniel MT, et al. Proposed revised criteria for the classification of acute myeloid leukemia a report of the French-American British cooperative Ann Intern Med. 1985; 103: 620-625.
- Arber DA, Orazi A, Hasserjian R, et The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016; 127: 2391.
- Schuler E, Schroeder M, Neukirchen J, et al. Refined medullary blast and white blood cell count based classification of chronic myelomonocytic leukemias. Leuk Res. 2014; 38: 1413-1419.
- Such E, Cervera J, Costa D, et al. Cytogenetic Risk Stratification in Chronic Myelomonocytic Haematologica. 2011; 96: 375-383.
- Djordjevic V, Jankovic G, Suvajdzic N, et A der(14)t(1;14) (q12;p11) in chronic myelomonocytic leukemia. Cancer Genetics and Cytogenetics. 2005; 160: 89-93.
- Itzykson R, Kosmider O, Renneville A, et Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol. 2013; 31: 2428-2436.
- Abdel-Wahab O, Pardanani A, Patel J, et al. Concomitant analysis of EZh3 and ASXL1 mutations in myelofibrosis chronic myelomonocytic leukemia and blast-phase myeloproliferative Leukemia. 2011; 25: 1200- 1202.
- Ernst T, Chase A, Zoi K, et Transcription factor mutations in myelodysplastic/myeloproliferative neoplasms. Haematologica. 2010; 95: 1473-1480.
- Gelsi-Boyer V, Trouplin V, Roquain J. ASXL1 mutation is associated with poor prognosis and acute transformation in chronic myelomonocytic leukaemia. Br J Haematol. 2010; 151: 365-375.
- Grossmann V, Kohlmann A, Eder C, et Molecular profiling of chronic myelomonocytic leukemia reveals diverse mutations in >80% of patients with TET2 and EZh3 being of high prognostic relevance. Leukemia. 2011; 25: 877-879.
- Tefferi A, Lim KH, Abdel-Wahab O, et Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia. 2009; 23: 1343-1345.
- Gelsi-Boyer V, Trouplin V, Adelaide J, et al. Genome profiling of chronic myelomonocytic leukemia frequent alterations of RAS and RUNX1 genes. BMC Cancer. 2008; 8:
- Kohlmann A, Grossmann V, Klein HU, et al. Next-generation sequencing technology reveals a characteristic pattern of molecular mutations in 72.8% of chronic myelomonocytic leukemia by detecting frequent alterations in TET2, CBL, RAS, and RUNX1. J Clin Oncol. 2010; 28: 3858-3865.
- Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera essential thrombocythemia and myeloid metaplasia with Cancer Cell. 2005; 7: 387-397.
- Makishima H, Cazzolli H, Szpurka H, et Mutations of e3 ubiquitin ligase cbl family members constitute a novel common pathogenic lesion in myeloid malignancies. J Clin Oncol. 2009; 27: 6109-6116.
- Daver N, Strati P, Jabbour E, et FLT3 mutations in myelodysplastic syndrome and chronic myelomonocytic leukemia. Am J Hematol. 2013; 88: 56-59.
- Laborde RR, Patnaik MM, Lasho TL, et SETBP1 mutations in 415 patients with primary myelofibrosis or chronic myelomonocytic leukemia: independent prognostic impact in CMML. Leukemia. 2013; 27: 2100-2102.
- Damm F, Itzykson R, Kosmider O, et al. SETBP1 mutations in 658 patients with myelodysplastic syndromes, chronic myelomonocytic leukemia and secondary acute myeloid Leukemia. 2013; 27: 1401-1403.
- Itzykson R, Solary E. An evolutionary perspective on chronic myelomonocytic leukemia. Leukemia. 2013; 27: 1441-1450.
- Patnaik MM, Itzykson R, Lasho TL, et ASXL1 and SETBP1 mutations and their prognostic contribution in chronic myelomonocytic leukemia a two center study of 466 patients. Leukemia. 2014; 28: 2206-2212.
- Patnaik MM, Wassie EA, Lasho TL, et Blast transformation in chronic myelomonocytic leukemia Risk factors genetic features survival and treatment outcome. Am J Hematol. 2015; 90: 411-416.
- Patnaik MM, Wassie EA, Padron E, et Chronic myelomonocytic leukemia in younger patients: molecular and cytogenetic predictors of survival and treatment outcome. Blood Cancer J. 2015; 5: e280.
- Vallapureddy R, Lasho TL, Hoversten K, et Nucleophosmin1 NPM1 mutations in chronic myelomonocytic leukemia and their prognostic relevance. Am J Hematol. 2017; 92: E614-E618.
- Patnaik MM, Pierola AA, Vallapureddy R, et al. Blast phase chronic myelomonocytic leukemia Mayo MDACC collaborative study of 171 Leukemia. 2018; 32: 2512-2518.
- Maeda T, Ashie T, Kikuiri K, et al. Chronic myelomonocytic leukemia with polyneuropathy and IgA-paraprotein. Jpn J 1989; 28: 709-716.
- Niscola P, Siniscalchi A, Tendas A, et Chronic myelomonocytic leukemia coexisting with monoclonal gammopathy: concomitant response to azacitidine of both disorders. Ann Hematol. 2015; 94: 1753-1754.
- Hagihara M, Inoue M, Kodama K, et Simultaneous manifestation of chronic myelomonocytic leukemia and multiple myeloma during treatment by prednisolone and eltrombopag for immune-mediated thrombocytopenic purpura. Case Rep Hematol. 2016; 2016: 4342820.
- Ferrara F, Del Vecchio L, Antonolfi I, et Chronic lymphocytic leukemia coexisting with chronic myelomonocytic leukemia. Haematologica. 1992; 77: 171-173.
- Gaulier A, Jary Bourguignat L, Serna R, et Occurrence of angioimmunoblastic T cell lymphoma in a patient with chronic myelomonocytic leukemia features. Leuk Lymphoma. 2000; 40: 197-204.
- Kouides PA, Bennett Transformation of Chronic Myelomonocytic Leukemia to Acute Lymphoblastic Leukemia Case Report and Review of the Literature of Lymphoblastic Transformation of Myelodysplastic Syndrome. Am J Hematol. 1995; 49: 157-162.
- Yamamoto M, Nakagawa M, Ichimura N, et Lymphoblastic Transformation of Chronic Myelomonocytic Leukemia in an Infant. Am J Hematol. 1996; 52: 212-214.
- Quivoron C, Couronné L, Della Valle V, et TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011; 20: 25-38.
- Lemonnier F, Couronné L, Parrens M, et al. Recurrent TET2 mutations in peripheral T cell lymphomas correlate with TFH- like features and adverse clinical parameters. Blood. 2012; 120: 1466-1469.
- Solary E, Bernard OA, Tefferi A, et The Ten-Eleven Translocation 2 TET2 genes in hematopoiesis and hematopoietic diseases. Leukemia. 2014; 28: 485-496.
- Silverman LR, Demakos EP, Peterson BL, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group J Clin Oncol. 2002; 20: 2429-2440.