Identification of Genetic Alterations in Prostate Cancer Using Gene Expression Profiling
Author'(s): Zeynep Kucukakcali* and Ipek Balikci Cicek
Department of Biostatistics and Medical Informatics, Inonu University Faculty of Medicine, 44280, Malatya, Turkey.
*Correspondence:
Zeynep KÜÇÜKAKÇALI, Res. Asst. Dr., Inonu University,Faculty of Medicine, Department of Biostatistics and Medical Informatics, Malatya, Turkey, Tel: +90 422 341 0660/1337.
Received: 02 Aug 2023; Accepted: 07 Sep 2023; Published: 15 Sep 2023
Citation: Kucukakcali Z, Cicek IB. Identification of Genetic Alterations in Prostate Cancer Using Gene Expression Profiling. Cancer Sci Res. 2023; 6(1): 1-7.
Abstract
Aim: In this study, an open-access dataset of mRNAs from primary prostate cancer samples and matched normal tissue samples was used to identify genetic alterations that play a role in the development of prostate cancer, the most common malignant tumor in men with increasing incidence.
Material and Methods: The dataset used in the study consists of 10 primary prostate cancer samples and a sample of normal tissue. 20024 genes were sequenced from these tissues. Gene expression analysis was performed with the data set using the limma package in the R programming language. Enrichment analysis was performed with the clusterProfiler package.
Results: 1028 of 20024 genes showed different up- or down-regulation in primary prostate cancer tissues compared to normal tissues. According to the results of the enrichment analysis with these genes, the processes that play a role in primary prostate cancer are protein binding, binding, cellular process, biological process, and response to stimulus.
Conclusion: As a result, it can be said that the genetic structure of primary prostate cancer tissues changes compared to normal tissues, with the information obtained from gene expression analysis and enrichment analysis. It can be said that it is possible to increase the therapeutic effectiveness of the disease by analyzing the biomarkers with comprehensive and various methods.
Keywords
Introduction
With an anticipated 1 276 000 new cancer cases and 359 000 deaths in 2018, prostate cancer is the second most often diagnosed disease and the sixth leading cause of cancer mortality among men globally. Because of population increase and aging, the global prostate cancer burden is anticipated to rise to over 2.3 million new cases and 740,000 deaths by 2040 [1]. While the most known risk factors for prostate cancer are family history, advanced age,and black race. In addition, some studies have shown that low plasma selenium and alpha-tocopherol concentration, individual characteristics such as obesity and height reached in adults, use of dairy products, and a diet high in calcium increase the risk of prostate cancer [2,3]. Prostate cancer can be asymptomatic in the early stages and has an indolent course, requiring little or no therapy. However, the most common complaint is difficulty urinating, increased frequency, and nocturia, all of which can be caused by prostatic enlargement. Because the axis skeleton is the most prevalent location of bone metastatic illness, more advanced stages of the disease may manifest with urine incontinence and back discomfort. Many prostate tumors are identified by increased plasmatic levels of prostate-specific antigen (PSA > 4 ng/mL), a glycoprotein that is routinely produced by prostate tissue. However, because men without cancer have been reported to have increased PSA levels, a tissue biopsy is the gold standard for confirming the existence of malignancy [4].
Many quantifiable characteristics of cancer cells differ, including proliferation, metastatic potential, and treatment resistance. Such variations are caused by heritable genetic and epigenetic changes [5,6]. Every patient's cancer has distinct genomic and phenotypic alterations, but a growing number of studies in recent years have emphasized the high amount of genetic and phenotypic heterogeneity within a specific tumor [6-8]. The hereditary risk of prostate cancer contributes significantly to tumor development. It is believed that around 20% of individuals with prostate cancer have a family history of the disease, which may arise not only from shared genes but also from a similar pattern of exposure to specific environmental carcinogens and related lifestyle choices [9,10]. Several studies have found that inherited genetic background is linked to an elevated risk of prostate cancer, accounting for roughly 5% of disease risks [11,12].
Many studies on prostate cancer are aimed at both identifying genes involved in the hereditary form of prostate cancer and identifying mutations that occur in the same acquired form . There is no evidence yet on how to prevent the disease, which has an increasing prevalence in developed countries and hereditary infrastructure, and there is a need for genomic studies that can reveal the cause of the disease and explain the genomic and molecular infrastructure associated with the disease. In conclusion, a comprehensive examination of prostate cancer epidemiology and risk factors and performing genetic studies can help to understand the relationship between genetic abnormalities and the function of the environment to generate disease-associated mutations and/or promote tumor growth.
In recent years, studies have focused on the development of new genetic technologies and comprehensive analyzes of genetic and epigenetic changes in prostate cancer. The aim of this study is to determine the genes that cause prostate cancer and which pathway these genes are associated with, using open-access mRNA profile data obtained between primary prostate cancer samples and matched normal tissues in order to determine the genetic mechanism of prostate cancer.
Material and Methods
Dataset features and acquisition of mRNA data
The open-access data set used in the study was obtained from The National Center for Biotechnology Information (NCBI) with the "GSE114740" GEO data code. Samples in the dataset include primary prostate cancer samples and matched normal tissues. mRNAs were obtained by Illumina HiSeq 2000 using deep sequencing from 10 primary prostate cancer samples and matched normal tissue.
Bioinformatics, gene expression, and enrichment analysis Bioinformatics is the collection, storage, organization, archiving, analysis, and presentation of results based on theory and practice in a discipline such as biology, medicine, behavioral, or health sciences. Furthermore, it is focused on the study and development of computational tools and methodologies to broaden the use and processing of data produced through studies or the application of recognized procedures. Obtained as a consequence of research or the use of well-known methodologies. Analyses in bioinformatics are performed by selecting a database and a program that allows bioinformatic analysis to be performed in line with the biological question, molecule, or structure to be analyzed. The data and findings acquired as a consequence of the analyses are combined and analyzed analytically in light of previously available information about the subject in the literatüre [13].
Changes in an organism's or cell's physiology will be accompanied by changes in the pattern of gene expression, making gene expression analysis important in many fields of biological study. The still-in-development DNA microarray method is used to study gene expression by hybridizing mRNA to a high-density array of immobilized target sequences, each corresponding to a distinct gene. The effect of chemicals on gene expression, for example, can reveal functional and toxicological qualities. Expression studies on clinical samples, both healthy and sick, may lead to the identification of novel biomarkers [14].
Although RNA expression analyses have become a routine tool in many studies, obtaining biological insights from the information obtained as a result of the analyzes and being able to correlate the genes obtained with the disease state remains a major challenge. The high number of genes obtained as a result of expression analysis and expressed (up or down) in the disease state causes them not to be fully associated with the disease molecularly. Therefore, genes with common characteristics should be considered together in order to make associations. Enrichment analysis is a process developed exactly from this approach. That is, while the enrichment analysis deals with genes; focuses on gene clusters, i.e. groups of genes that share common biological functions, chromosomal location, or arrangements. Thus, the molecular processes of the disease can be understood more easily by obtaining the structures formed by the genes acting together [15].
Bioinformatics analysis
Gene expression analyses were done on mRNA data collected from primary prostate cancer samples and matched normal tissues in this investigation. In the investigation, the limma package, which is accessible in the R programming language and permits expression analysis, was employed [16]. Limma (Linear Models for Microarray Analysis) is a library for evaluating gene expression microarray data, with a focus on the use of linear models for assessing specified experiments and determining differential expression. The packet's functionalities are applicable to all gene expression methods, such as microarrays, RNA-seq, and quantitative PCR. The results are presented in the form of a table of genes sorted in order of relevance and a graph illustrating differentially expressed genes. The table of findings contains corrected P and log2-fold change (log2FC) values, with genes with the lowest p values being the most trustworthy. Log2FC>1 and p<0.05 were used to identify up-regulated genes, whereas log2FC-1 and p<0.05 were used to identify down-regulated genes. The volcano graph was utilized in the study to illustrate differentially expressed genes. In the volcano plot, genes with upregulation between cancerous tissue and normal tissue are shown in red, genes with downregulation in blue, and genes that do not show differentiation in both tissues are shown in black. The clusterProfiler package in R was used for the enrichment analysis with the results obtained from the gene expression analysis [17]. The results are given with dotplot, enrichment map and category netplot, where we can see the common functional mechanisms of genes.
Results
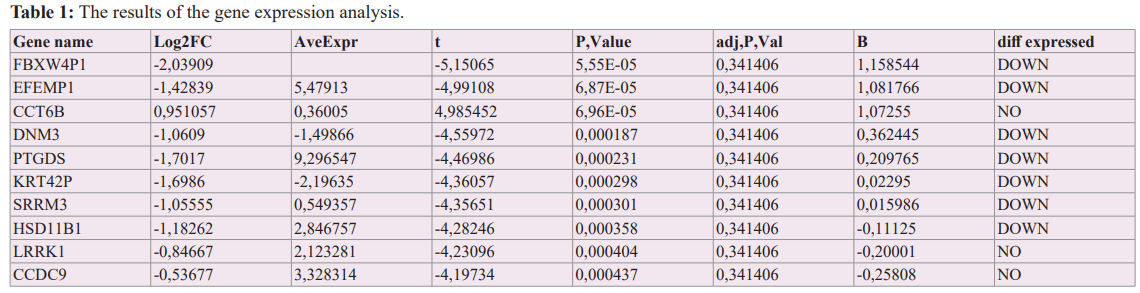
20024 genes were sequenced in the study. As a result of the gene expression analysis, while 302 genes were up-regulated, 726 genes were down-regulated. Among these genes, 10 genes that can be said to be the most important associated with the disease according to the results obtained by ranking according to the p-value are given in Table 1.
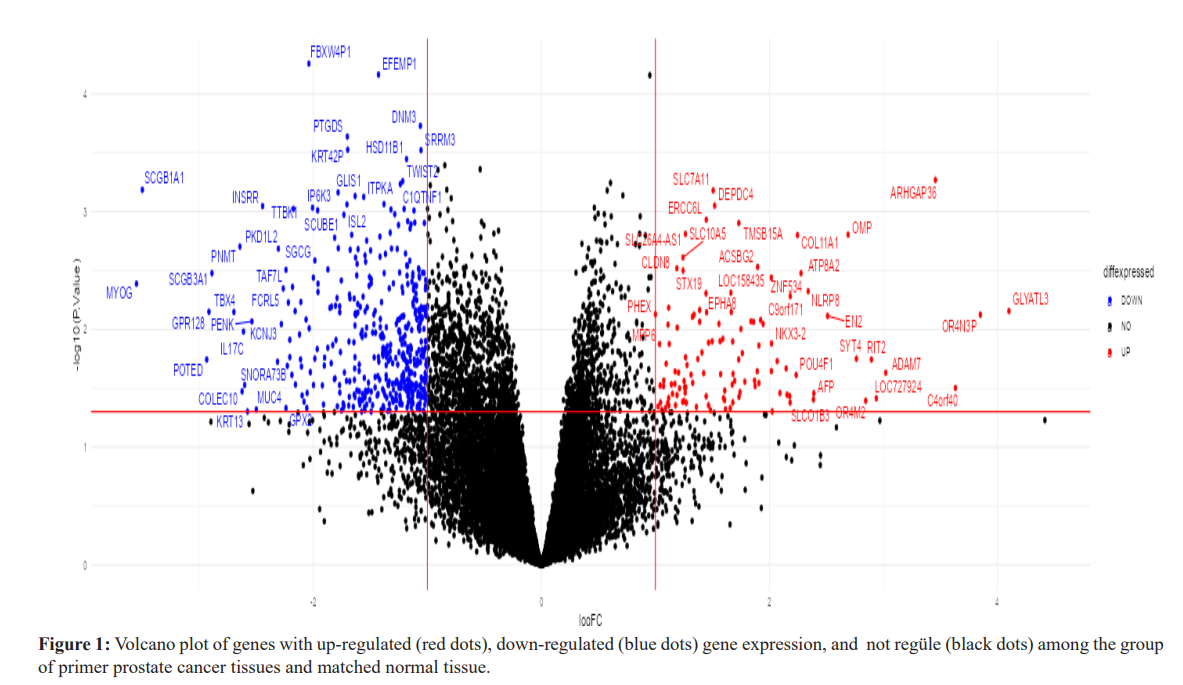
The genes that were most up-regulated in primary prostate cancer tissue compared to the matched normal tissue were OR4Q3 (Log2FC=4.41), GLYATL3 (Log2FC=4.10), OR4N3P (Log2FC=3.84), C4orf40 (Log2FC=3.63), ARHGAP36 (Log2FC=3. 45), ADAM7 (Log2FC=3.02), LOC100129636 (Log2FC=2.96), LOC727924 (Log2FC=2.93), RIT2(Log2FC=2.89), and OR4M2 (Log2FC=2.84). The genes that were most down-regulated in primary prostate cancer tissue compared to the matched normal tissue were MYOG (Log2FC=3.55), SCGB1A1 (Log2FC=3.50), POTED (Log2FC=2.93), GPR128 (Log2FC=2.91), BMP5 (Log2FC=2.90), SCGB3A1 (Log2FC=2.89), TBX4 (Log2FC=2.69), PNMT (Log2FC=2.64), COLEC10 (Log2FC=2.62), and IL17C (Log2FC=2.61). The volcano plot used to visualize differentially expressed genes is given in Figure 1.
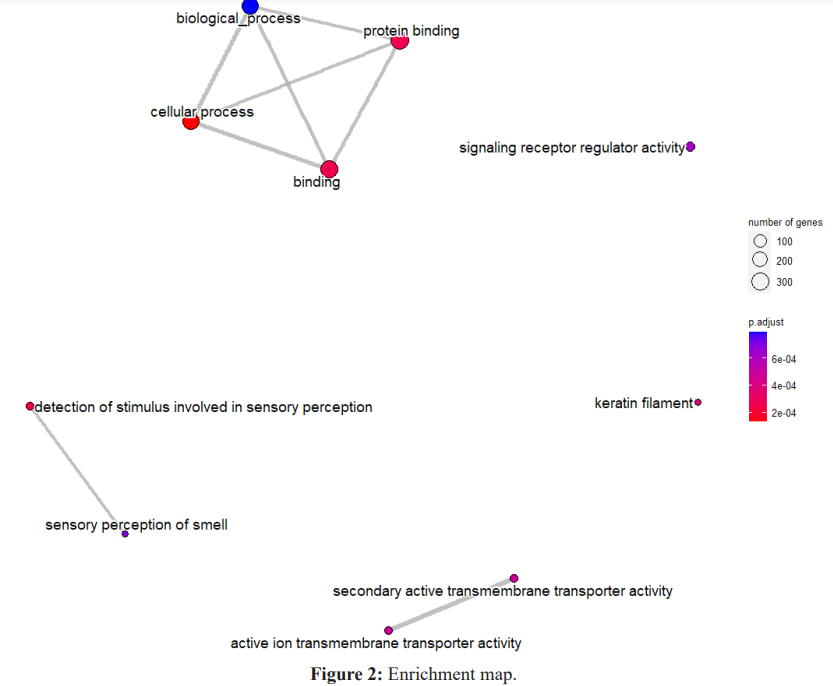
According to the results of the enrichment analysis, the metabolic mechanisms associated with the disease are presented in Figure 2 and Figure 3
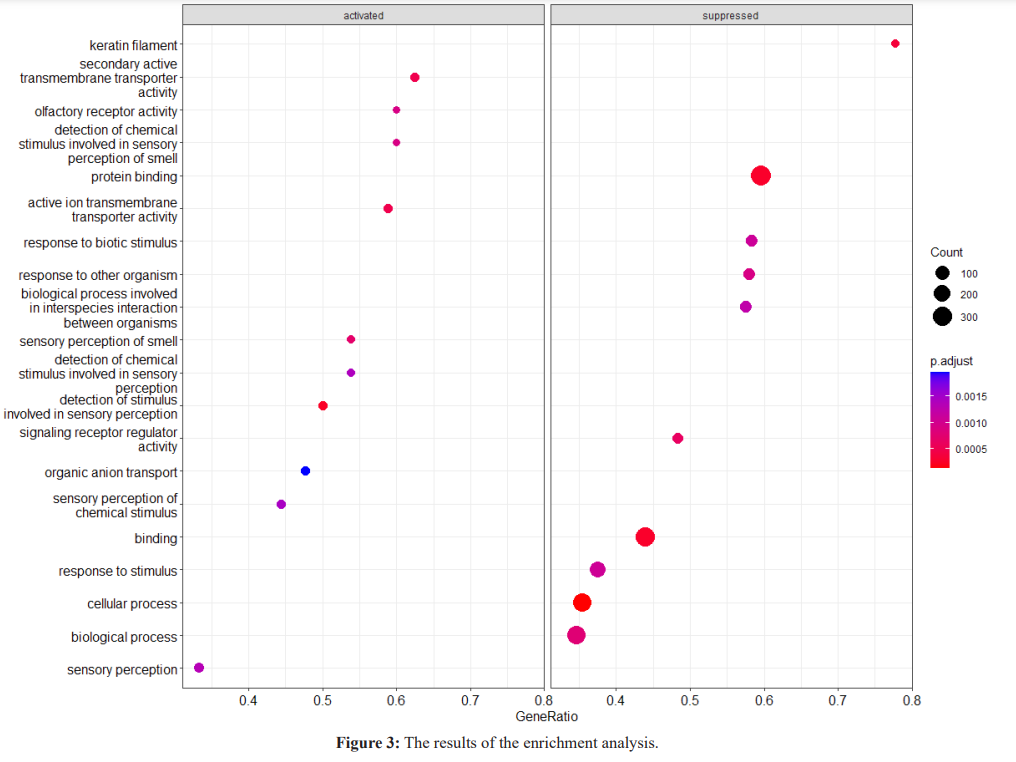
The graphic in which we can see the relations of genes with metabolic mechanisms in detail is given in figure 4. This graph shows the processes most affected by the disease state and the genes that may be important to the processes.
Discussion
Prostate cancer is one of the most malignant tumors in men, and its incidence has increased in recent years [18]. Thanks to the advances in medicine in recent years, there has been a significant increase in the survival and prognosis of patients with prostate cancer, especially in developed countries. While the 5-year survival rate was 66.9% in 1975, this rate increased to 97.8% in 2016, indicating the progression trend in prostate cancer [19]. Genetic and epigenetic factors play a very important role in the development of prostate cancer [18]. The most commonly used biomarker in the diagnosis of prostate cancer is PSA, and this method has low specificity and cannot detect the disease completely. Biopsy, which is an invasive method and the gold standard for prostate diagnosis, causes great pain to patients. For this reason, early diagnosis and prognostic markers that can be used in the detection of prostate cancer and that will benefit the well-being of patients are important [20]. Therefore, benefiting from bioinformatics and genomic studies has become very important in terms of the detection of the disease.
Microarrays and high-throughput sequencing technologies have been widely employed in screening biomarkers for cancer diagnosis, therapy, and prognosis, and have played an essential role in the research of the occurrence and development of cancer. At the same time, the integrated bioinformatics technique is employed in cancer research to overcome the limitations of diverse technological platforms or limited sample size. The combined use of numerous databases can integrate data from different independent research and gather more clinical samples for data mining, resulting in more robust and reliable analysis. These new bioinformatics methods offer enormous potential for the progress of cancer biomarker research [20].
New genetic technologies developed in recent years have enabled the comprehensive analysis of changes in prostate cancer, which has a genetic and epigenetic background. Thus, targeted functional studies were processed with the obtained information, enabling the revealing of critical genes that play a role in the initiation and progression of prostate cancer, and the critical metabolic mechanisms and signaling pathways associated with them [4]. This information obtained will allow the development of new approaches to be able to make therapeutic interventions in the light of new studies. And the prognosis of the disease and treatment strategies will be fully determined. In order to achieve such goals, researchers have given importance to genetic studies that may affect the development of prostate cancer and are examining genetic changes.
Therefore, this study aims to identify the processes and genes involved in the development of prostate cancer using an open- access dataset of mRNA data. For this purpose, the dataset used in the study consists of 10 primary prostate cancer tissues and matched normal tissue samples. 20024 genes were sequenced in the data set. According to the results of the bioinformatics analysis, 1028 genes showed different regulation. The OR4Q3 gene showed 21.25 fold up-regulation in primer prostate cancer samples compared to normal tissue samples. Likewise, the GLYATL3, OR4N3P, C4orf40, ARHGAP36, ADAM7, LOC100129636, LOC727924, RIT2, and OR4M2 genes had up-regulated gene expression of 17.14, 14.32, 12.38, 10.92, 8.11, 7.78, 7.62, 7.41,and 7.16 fold, respectively. Moreover, the MYOG gene showed 11.71-fold down-regulation in pancreatic ductal adenocarcinoma samples compared to normal tissue samples. Likewise, the SCGB1A1, POTED, GPR128, BMP5, SCGB3A1, TBX4, PNMT,COLEC10, and IL17C genes had down-regulated gene expression of 11.31, 7.62, 7.51, 7.46, 7.41, 6.45, 6.23, 6.14, and 6.10 fold,respectively.
According to the results of the enrichment analysis performed by considering genes showing different regulation, protein binding, bindng, cellular process, biological process, responce to stimulus processes were obtained as important processes.
As a result, it can be said that the genetic structure of primary prostate cancer changes with the information obtained from gene expression analysis and enrichment analysis, and it may be possible to increase the therapeutic efficacy of the disease

with comprehensive and various genetic studies to be carried out considering these changes. With the biomarkers used in the diagnosis of the disease, appropriate treatment for primary prostate cancer can be developed and the disease can be treated before it progresses. With the new oncological treatment selection developed in this way, the treatment can reach the target and mortality rates can be reduced.
References
- Culp MB, Soerjomataram I, Efstathiou JA, et Recent Global Patterns in Prostate Cancer Incidence and Mortality Rates. European urology. 2020; 77: 38-52.
- Mayne S, Morse D, Winn D, et al. Cancer epidemiology and Oxford University Press New York. 2006; 674- 696.
- Hayes RB, Ziegler RG, Gridley G, et al. Dietary factors and risks for prostate cancer among blacks and whites in the United Cancer Epidemiology Biomarkers & Prevention. 1999; 8: 25-34.
- Rawla P. Epidemiology of prostate cancer. World journal of 2019; 10: 63.
- Alizadeh AA, Aranda V, Bardelli A, et Toward understanding and exploiting tumor heterogeneity. Nature medicine. 2015; 21: 846-853.
- MarusykA,Almendro V, Polyak Intra-tumour heterogeneity: a looking glass for cancer? Nature reviews Cancer. 2012; 12: 323-334.
- Gerlinger M, Catto JW, Orntoft TF, et Intratumour heterogeneity in urologic cancers: from molecular evidence to clinical implications. European Urology. 2015; 67: 729-37.
- Mitchell T, Neal The genomic evolution of human prostate cancer. British journal of cancer. 2015; 113: 193-198.
- Gallagher RP, Fleshner N. Prostate cancer: 3. Individual risk CMAJ: Canadian Medical Association Journal. 1998; 159: 807-813.
- Mohler JL, Armstrong AJ, Bahnson RR, et Prostate cancer, version 1. Journal of the National Comprehensive Cancer Network. 2016; 14: 19-30.
- Ferrís-i-Tortajada J, García-i-Castell J, Berbel-Tornero O, et Constitutional risk factors in prostate cancer. Actas urologicas espanolas. 2011; 35: 282-288.
- Sridhar G, Masho SW, Adera T, et al. Association between family history of cancers and risk of prostate cancer. Journal of Men's 2010; 7: 45-54.
- Akalin Introduction to bioinformatics. Molecular nutrition & food research. 2006; 50: 610-619.
- Van Hal NL, Vorst O, van Houwelingen AM, et The application of DNA microarrays in gene expression analysis. Journal of biotechnology. 2000; 78: 271-280.
- Subramanian A, Tamayo P, Mootha VK, et Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102: 15545-15550.
- Smyth Limma: linear models for microarray data. Bioinformatics and computational biology solutions using R and Bioconductor: Springer. 2005; p. 397-420.
- Wu T, Hu E, Xu S, et al. Cluster Profiler 4.0: A universal enrichment tool for interpreting omics The innovation. 2021; 2.
- Mirzaei S, Paskeh MDA, Okina E, et Molecular Landscape of LncRNAs in Prostate Cancer: A focus on pathways and therapeutic targets for intervention. Journal of Experimental & Clinical Cancer Research. 2022; 41: 214.
- Races A, Males MWMB. SEER Cancer Statistics Review 1975-2017. 2020.
- Liu S, Wang W, Zhao Y, et al. Identification of Potential Key Genes for Pathogenesis and Prognosis in Prostate Cancer by Integrated Analysis of Gene Expression Profiles and the Cancer Genome Frontiers in oncology. 2020; 10: 809.