Primary Pulmonary Histiocytic Sarcoma Presenting with Hemoptysis
Author'(s): Emily Bryer DO*, Megan Manno DO, Paul Kinniry MD, Lee Hartner MD
Pennsylvania Hospital, University of Pennsylvania, US.
*Correspondence:
Emily Bryer DO, Pennsylvania Hospital, University of Pennsylvania,US.
Received: 27 January 2019; Accepted: 20 February 2019
Citation: Emily Bryer DO, Megan Manno DO, Paul Kinniry MD, et al. Primary Pulmonary Histiocytic Sarcoma Presenting with Hemoptysis. Cancer Sci Res. 2019; 2(1); 1-5.
Abstract
Histiocytic sarcomas are malignant tumors of tissue macrophages that comprise an incredibly rare group of hematopoietic neoplasms. Investigation of historical diagnoses of histiocytic sarcoma reveals misclassifications of B-cell neoplasms and anaplastic large cell lymphomas, making the true diagnosis of histiocytic sarcoma even more rare than previously recognized. Histiocytic sarcomas are diagnosed by histology, morphology, and immunohistochemistry. They often manifest with extranodal presentations that are typically cutaneous or gastrointestinal in origin. We present the fifth documented case of primary pulmonary histiocytic sarcoma that developed in a 61-year-old man and manifested as hemoptysis with interval development of hemothorax. Prognosis is poor and the rarity of histiocytic sarcomas preclude the establishment of standardized treatment guidelines.
Keywords
Case Report
A 61-year-old man with a past medical history significant for generalized anxiety disorder, hypertension, hyperlipidemia, and a provoked deep venous thrombosis 20 years prior presented to an outside hospital with progressive dyspnea on exertion, left-sided pleuritic chest pain, and one episode of hemoptysis. Over the year prior to presentation, he reported increased non-productive cough, difficulty balancing, and non-specific weakness. Review of systems was also positive for subjective fevers, night sweats, and a 30-pound unintentional weight loss over the previous year. Social history was remarkable for current tobacco use with a 35-pack year history smoking one pack per day and drinking one can of beer daily. He had long-standing marijuana use that stopped 6 months prior to presentation. The patient was a former army mechanic and worked with brake linings. Family history significant for a father who died of emphysema in his 50’s and a mother with coronary artery disease and diabetes.
Upon arrival to outside hospital, EKG was normal sinus rhythm at 78bpm. Chest x-ray revealed a large opacity in the left upper lung. Computed tomography (CT) scan of the chest showed a large 10.5x6.3x8.3cm left upper lung mass that was inseparable from the left side of the aortic arch and extended to the left suprahilar region. There were severe emphysematous changes, a right adrenal mass, a 1cm hypodense lesion in the posterior right thyroid lobe, and large lymphadenopathy in the middle mediastinum and hilar regions. Magnetic resonance imaging (MRI) of the brain revealed multiple regions of dural enhancement concerning for metastases, but no discrete parenchymal metastases. The patient had a CT-guided left lung biopsy that was positive for the following immunohistochemistry stains: CD68, CD163, vimentin, CD10, CD4, EMA (focal weak), AE1/AE3 (focal), panCK (focal). The biopsy was negative for lysozyme, TTF1, napsin A, p63, CK7, CK20, arginase, PAX8, inhibin, calretinin, CD34, Mart1, HMB45, synaptophysin, chromogranin, CD1a, CD56, SALL4, CD45, myeloperoxidase, CD15, desmin, SMA, CD3, CD20, CD117, CD30, CD21, CD23, CAM5.2, S100 and ALK. The Ki-67 proliferative index is approximately 50%. H&E revealed diffuse proliferation of large pleomorphic cells. The cells were large with abundant cytoplasm, eccentrically located nuclei with prominent nucleoli, and vesicular chromatin.
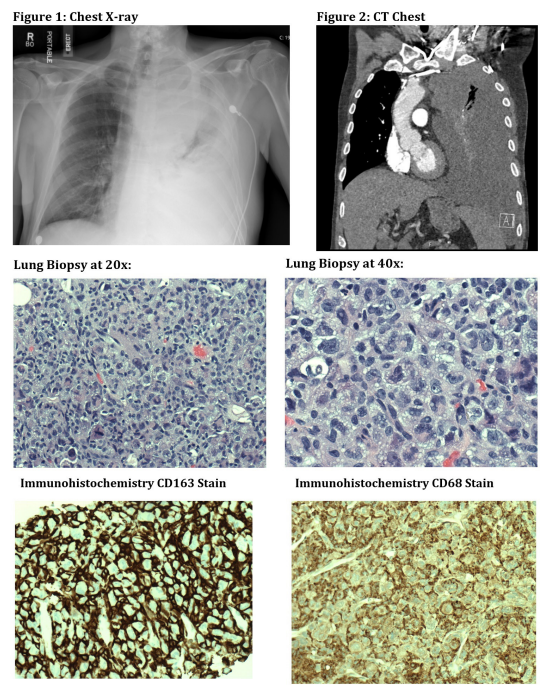
The post-biopsy chest x-ray demonstrated a moderate to large partially loculated pleural fluid collection. The patient was discharged home and presented to our hospital four days later with near-syncope. Vital signs on admission were remarkable for pulse 86bpm, blood pressure 104/70mmHg, temperature 97.7F, respiratory rate was 20 and SpO2 was 86% on room air. He became more tachypneic with a respiratory rate of 28 and he was placed on a non-rebreather with improvement of his SpO2 to 100%. Labs on admission were remarkable for WBC 39.6, hemoglobin 11.1, platelets 452, INR 1.2, D-dimer 7.06, LDH 317. Transthoracic echocardiogram revealed an ejection fraction of 75% without any wall motion abnormalities. CXR on admission (Figure 1) revealed near complete opacification of the left hemithorax with apparent mass effect in the left mediastinum concerning for neoplasm. A CT (Figure 2) was obtained and demonstrated a large loculated pleural effusion with soft tissue pleural masses concerning for malignancy. There was interval development of previously unilateral adrenal metastasis to bilateral adrenal metastases. The patient underwent a thoracentesis which yielded 1.4L of hemorrhagic fluid. This was found to be an exudative effusion (pH 7.2, leukocytes 6414, erythrocytes 889,000, LDH 1090, amylase 14, glucose 43, albumin 2.3, protein 3.9). Fluid cultures were negative and flow cytometry of the left pleural fluid did not reveal any evidence of a hematolymphoid malignancy. Over the next few days, he developed hypoxia to 70% on 4L O2 and was transferred to the Intensive Care Unit. A chest tube was urgently placed with 1.6L output of hemorrhagic fluid. Bronchoscopy revealed left mainstem bronchus with complete occlusion and thick purulent secretions. Some fullness and extrinsic compression of the left lower lobe without endobronchial lesion was also seen. He subsequently developed severe post-obstructive pneumonia, acute hypoxic respiratory failure requiring intubation, and septic shock requiring vasopressors. White blood cell count increased to 104.2 (from 39.6). Due to his functional status, he was not a candidate for cytotoxic chemotherapy and he died a few days after admission.
Histiocytic Sarcoma
Histiocytic sarcoma is an incredibly rare and aggressive malignant neoplasm. It is derived from monocytic and macrophage lineage and has uncertain molecular pathogenesis [1,2]. Histiocytic sarcoma displays the morphological and immunophenotypic features of mature tissue histiocytes, also known as tissue macrophages [3,4]. Histiocytic sarcoma comprises less than 0.5% of all Non-Hodgkin’s lymphoma [5,6]. Most patients are adult men with median age ranging between 46 and 55 [2,7]. Histiocytic sarcoma is associated with a poor prognosis with about 60% of cases metastatic on presentation [3]. The clinical presentation of histiocytic sarcoma is highly variable and can range from isolated lymph node involvement to disseminated extra-nodal disease [8]. It typically manifests in the lymph nodes, skin, and the gastrointestinal tract [4]. Symptoms include fever, weight loss, and night sweats as well as organ-specific manifestations such as hepatosplenomegaly and cutaneous lesions [9]. The diagnosis of histiocytic sarcoma is complex due to the mimicry of other lymphoid tissue malignancies in both clinical presentation and morphological appearance [2]. Furthermore, the characteristic histiocytic markers are not commonly included in the routine diagnostic panel of antibodies used to investigate undifferentiated neoplasms [10].
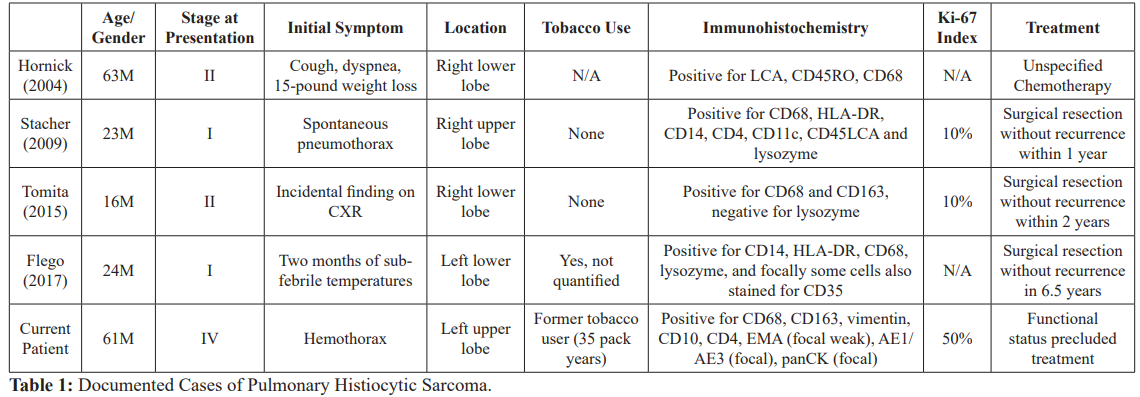
Over the past few decades, advances in immunohistochemistry have changed the identification and classification of histiocytic sarcoma. Due to these developments and identification of tumor-specific cell markers, many previously diagnosed histiocytic sarcomas are now recognized as misdiagnosed Non-Hodgkin lymphomas, specifically diffuse and anaplastic B-cell lymphoma, peripheral T-cell lymphoma associated with hemophagocytic syndrome, and lymphoma with associated reactive macrophages [4,11]. Historical inconsistencies in terminology and diagnostic criteria of these lesions complicate their diagnosis and characterization [1]. The diagnosis of histiocytic sarcoma is established by histological, immunohistochemical, and morphological analysis. The differential diagnosis of histiocytic sarcoma includes other tumors of dendritic or mononuclear phagocyte origin such as Langerhans cell sarcoma, interdigitating dendritic cell sarcoma and follicular dendritic cell sarcoma [12]. While histiocytic sarcomas may develop sporadically, they may also be related to a synchronous or metachronous hematologic malignancy [3]. Transdifferentiaton of follicular lymphomas and acute lymphoblastic leukemia have both been described with histiocytic sarcoma [3,13]. There may be a cooperative interaction between PTEN and p16(INK4A)/ p14(ARF) in the development of histiocytic sarcoma [2,4]. While only four cases of pulmonary histiocytic sarcoma have been identified, it has been more commonly described in the central nervous system, spleen, liver, and skin. Table 1 includes all four previously reported cases of pulmonary histiocytic sarcoma [7,14- 16].
Histiocytic sarcoma histology features large cells with rich eosinophilic cytoplasm that resemble epithelioid histiocytes [14]. These neoplasms usually have large, vesicular nuclei, prominent nucleoli and giant multinucleated cells [14]. While morphologically
histiocytic sarcomas may resemble other lymphoid malignancies, immunohistochemical staining helps to distinguish it from other neoplasms [2]. Historically, neoplasms have been misclassified as histiocytic in etiology due to histologic features and insufficient phenotypic data [1]. CD163, a hemoglobin scavenger receptor, is a recently identified marker that is specific to histiocytic lineage and has augmented the accuracy of histiocytic sarcoma diagnosis [1]. While CD68 is also a marker of histiocytic differentiation, it is less specific than CD163 and can also stain positive in carcinoma and melanoma [13]. In addition to CD163 and CD68, immunohistochemical stains of histiocytic sarcoma typically show cells that are reactive with neuron-specific enolase (NSE) and vimentin which are all typical of normal monocytes and histiocytes [2]. A non-reactive CD1a immunohistochemical stain differentiates histiocytic sarcoma from Langerhan’s histiocytosis [2].
Although disease stage and tumor size at presentation are two strong predictors of histiocytic sarcoma prognosis, the tumor is especially aggressive and often treatment-resistant [11,17]. Most patients present with advanced disease and succumb to progressive disease within two years of diagnosis [11,18]. There are no standardized guidelines for the optimal treatment of histiocytic sarcoma. Reported treatments of localized histiocytic sarcoma include surgery and radiation, while chemotherapy is reserved for advanced or metastatic disease. Chemotherapy with cyclophosphamide, doxorubicin, vincristine and prednisone (CHOP) has been reported as first-line therapy in patients with advanced histiocytic sarcoma [7]. Combination ifosfamide, carboplatin and etoposide (ICE) chemotherapy has also been trialed in patients with histiocytic sarcoma [3]. Patients with histiocytic sarcoma refractory to ICE chemotherapy have received trials of carboplatin/paclitaxel without significant response [3].
Due to the unknown molecular etiology of histiocytic sarcoma, targeted therapeutics have not previously been feasible [4]. Over the past few years, targeted therapies with imatinib, sorafenib, and bevacizumab have provided some encouraging results in the treatment of histiocytic sarcoma [19,20]. Treatment with Trametinib, a MEK inhibitor, has been successfully implemented as a treatment for histiocytic sarcoma of a MAP2K1 pathogenic variant that transformed from Rosai-Dorfman disease, a rare non- Langerhans cell histiocytosis [21]. The identification of CD52, an antigen expressed on many histiocytic cells, has translated into therapy with alemtuzumab, an anti-CD52 monoclonal antibody. Although it has only been documented in the pediatric population, the use of alemtuzumab in children with multi-organ involvement from histiocytic sarcoma that was refractory to aggressive conventional chemotherapy resulted in complete remission [20].
Case Discussion
Accurate identification of a pulmonary histiocytic sarcoma is rare. Sarcomas are of mesenchymal origin, and very rarely present as primary pulmonary tumors [22]. The presentation of this patient with hemoptysis and development of unilateral hemothorax with tracheal deviation is unique, even for a primary pulmonary tumor. While pulmonary sarcomas are rare, pulmonary histiocytic sarcomas are even more rare and we describe the fifth case reported to date. Compared with the four other previously documented cases, this case displays the most advanced disease at presentation and appeared to arise de novo without any other preceding or concomitant tumor. While tobacco use was only documented for one other previously reported case, our patient’s significant and current tobacco use may have contributed to the development of histiocytic sarcoma. It is unclear whether or not the patient’s father’s tobacco uses and pulmonary disease had a genetic and or environmental influence on this patient’s pulmonary disease or sarcoma development. Although working with brake linings has been associated with airborne asbestos [23], it is unknown whether or not this contributed to his histiocytic sarcoma and or the soft tissue pleural masses that he developed.
Immunohistochemistry stains positive for CD68 and CD163 were also found in other cases of pulmonary histiocytic sarcoma and, along with the morphology of the biopsy, confirm the rare diagnosis of histiocytic sarcoma. The presence of a right unilateral adrenal metastasis at the outside hospital with progression to bilateral adrenal metastases four days later emphasizes the virulence of histiocytic sarcoma. Furthermore, the presence of a solitary adrenal metastasis is typically ipsilateral to the laterality of the lung cancer [24]. This case displays a left-sided pulmonary mass with an initially contralateral solitary metastasis that rapidly progressed to bilateral adrenal metastases. While the mechanism of adrenal metastases in lung cancer is controversial, recent mathematical models suggest a lymphatic spread over hematogenous transmission [24].
Future Directions
Risk factors, implicated genes, and environmental exposures that increase the risk of histiocytic sarcoma are yet to be determined. While tobacco use has been correlated with the development of other pulmonary histiocytic disorders including Langerhans cell histiocytosis, further investigations are needed to determine if there is a relationship with tobacco and pulmonary histiocytic sarcoma. As our comprehension of the molecular pathogenesis of histiocytic sarcoma expands, greater opportunities for targeted treatments are anticipated. Although patients with central nervous system sarcomas have responded well to vemurafenib, a B-RAF inhibitor [25], its role in pulmonary histiocytic sarcomas is not yet clear. Additionally, PDL1 expression by histiocytic sarcomas suggest a potential role for therapy with PDL1 inhibitors [26]. Interestingly, the frequency of histiocytic sarcoma is much higher in dogs which may serve as a translational model system for future therapies [27]. Tumors suppressor genes PTEN and INK4A have distinct and synergistic roles in hematopoietic lineage development and play an integral role in the development of histiocytic sarcoma [4]. PI3K, RAS/MAPK, and cdk4/6 inhibitors may also serve as functional therapies for this currently intractable disease [4].
Conclusion
We describe an unusual presentation of an extremely rare malignancy. While there have only been four prior reported primary pulmonary histiocytic sarcomas documented to date, this case represents the highest Ki-67 index reported to date as well as the only case of pulmonary histiocytic sarcoma to present at Stage IV and with hemothorax. Furthermore, the diagnosis of a primary pulmonary sarcoma is unique in itself, as the cells are of mesenchymal origin. The rarity of pulmonary histiocytic sarcomas challenges the adoption of treatment guidelines. Unfortunately, this patient was critically ill upon presentation and was not a candidate for any intervention. Further research is needed to clarify optimal therapeutics in pulmonary histiocytic sarcoma as well as to elucidate the role of tobacco use and other risk factors on the development of pulmonary histiocytic sarcoma.
Acknowledgment
Dr. Ying Lu of Hahnemann Department of Pathology for images and captions.
References
- Vos JA, Abbondanzo SL, Barekman CL, et al. Histiocytic sarcoma a study of five cases including the histiocyte marker CD163. Modern Pathology. 2005; 18: 693-704.
- Alexiev BA, Sailey CJ, McClure SA, et Primary histiocytic sarcoma arising in the head and neck with predominant spindle cell component. Diagnostic Pathology. 2007; 2: 7.
- Kayikcioglu E, Aydin AA, Onder AF, et An extremely rare neoplasm, histiocytic sarcoma A report of two cases with an aggressive clinical course. Journal of Oncological Sciences. 2017; 3: 84-86.
- Carrasco DR, Fenton T, Sukhdeo, et al. The PTEN and INK4A/ARF tumor suppressors maintain myelolymphoid homeostasis and cooperate to constrain histiocytic sarcoma development in humans. Cancer Cell. 2006; 9: 379-390.
- Grogan TM, Pileri SA, Chan JKC, et al, Fletcher CDM Histiocytic sarcoma. In World Health Organization Classification of Tumours Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon ARC Press. 2008; 356-357.
- Pileri SA, Grogan TM, Harris NL, et Tumours of histiocytes and accessory dendritic cells an immunohistochemical approach to classification from the International Lymphoma Study Group based on 61 cases. Histopathology. 2002; 41:1-29.
- Hornick JL, Jaffe ES, Fletcher CD. Extranodal histiocytic sarcoma clinicopathologic analysis of 14 cases of a rare epithelioid Am J Surg Pathol. 2004; 28: 1133-1144.
- Zhao J, Niu X, Wang Z, et al. Histiocytic sarcoma combined with acute monocytic leukemia a case report. Diagnostic Pathology. 2015; 10: 110.
- Abu-Sanad A, Warsi A, Michael R, et al. Long-term remission after autologous stem-cell transplantation for relapsed histiocytic sarcoma. Current Oncology. 2012; 19: 289-291.
- El-Tatawy R, Mansour L, Gawish K, et Histiocytic sarcoma histiologic and immuno-phenotypic analysis a case study. 2008. Egyptian Dermatology Online Journal. 2008; 4: 1-9.
- Takahashi E, Nakamura S. Histiocytic sarcoma An updated literature review based on the 2008 WHO Classification. J Clin Exp Hematop. 2013; 53: 1-8.
- Windebank K, Visser J, Nanduri V. Advances in the understanding and management of histiocytic disorder. Pediatrics and Child Health. 2015; 26: 73-80.
- Skala SL, Lucas DR, Dewar R. Histiocytic Sarcoma. Arch Pathol Lab Med. 2018; 142: 1322-1329.
- Flego V, Popper H, Volaric D, et al. Pulmonary histiocytic sarcoma A case report and a literature review. American Journal of Internal Medicine. 2017; 5: 91-94.
- Stacher E, Beham-Schmid C, Terpe HJ, et al. Pulmonary histiocytic sarcoma mimicking pulmonary Langerhans cell histiocytosis in a young adult presenting with spontaneous pneumothorax a potential diagnostic pitfall. Virchows Arch. 2009; 455: 187-190.
- Tomita S, Ogura G, Inomoto, et al. Histiocytic sarcoma originating in the lung in a 16 year-old male. J Clin Exp Hematop. 2015; 55: 45-49.
- World Health Organization Classification of Tumors: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissue. Lyon France IARC Press. 2001; 274-289.
- Accurso K, McNamara W, Marsh C, et A case report of an extranodal histiocytic sarcoma presenting in the oral cavity. Oral surgery oral medicine oral pathology oral radiology and endodontology. 2010; 110: 681-810.
- Schlick K, Aigelsreiter A, Pichler M, et al. Histiocytic sarcoma- targeted therapy novel therapeutic options A series of 4 cases. Onkologie. 2012; 35: 447-450.
- Shukla N, Kobos R, Renaud T, et al. Successful treatment of refractory metastatic histiocytic sarcoma with alemtuzumab. Cancer. 2012; 118: 3719-3724.
- Kumamoto T, Aoki Y, Sonoda T, et al. A case of recurrent histiocytic sarcoma with MAP2K1 pathogenic variant treated with the MEK inhibitor trametinib. International Journal of Hematology. 2019; 109: 228-232.
- Alvina Y, Mallapasi MN, Rosie R, et al. Malignant fibrous histiocytoma in lungs a case report. American Journal of Medical Case Reports. 2017; 5: 217-220.
- Lemen RA. Asbestos in brakes exposure and risk of disease. American Journal of Industrial 2004; 45: 229-237.
- Bazhenova L, Newton P Mason J, Bethel K, et al. J Thorac Oncol. 2014; 9: 442-446.
- Idbaih A, Mokhtari K, Emile JF, et al. Dramatic response of a BRAF V600E-mutated primary CNS histiocytic sarcoma to vemurafenib. Neurology. 2014; 83: 1478-1480.
- Facchetti F, Pileri SA, Lorenzi L, et al. Histiocytic and dendritic cell neoplasms what have we learnt by studying 67 cases. Virchows Arch. 2017; 471: 467-468.
- Takada M, Hix J, Corner, et al. Targeting MEK in a translational model of histiocytic Molecular Cancer Therapeutics. 2018; 17: 2439-2450.