Therapeutic Apheresis and Immunosuppression in Immunologic Diseases: A Review and Own Observations
Author'(s): Rolf Bambauer1* and Ralf Schiel2
1 Formerly at the Institute for Blood Purification, Homburg/ Saar, Germany.
2 Clinic for Metabolic Diseases, Medigreif Inselklinik, Heringsdorf, Seeheilbad Heringsdorf, Germany.
*Correspondence:
Rolf Bambauer, MD, PhD., Frankenstrasse 4, 66424 Homburg, Germany, Tel.: 0049(0)6841-68500; Fax: 0049(0)6941-689561.
Received: 19 October 2021; Accepted: 11 November 2021
Citation: Bambauer R, Schiel R. Therapeutic Apheresis and Immunosuppression in Immunologic Diseases: A Review and Own Observations. Clin Immunol Res. 2021; 5(2): 1-36.
Abstract
Autoimmune diseases based on an immune pathogenesis produce autoantibodies and circulating immune complexes, which cause inflammation in the tissues of various organs. In most cases, these diseases have a bad prognosis without treatment, and the treatment is complicated in these diseases. With the introduction of therapeutic apheresis (TA), since more than 45 years, has led in combination with immunosuppressive therapies to a steady increase in survival rates over the last decades. TA is accepted as supportive therapy in all antibodymediated diseases. Further modern therapy are different human monoclonal antibodies (HMA) with or without TA. Besides, the pathological aspects, the first-line and second-line therapies for immunologic diseases, such as renal, neurological, hematological, dermatological, and autoimmune diseases, which are possible, are shown. For immunological diseases that can be treated with TA, the guidelines of the Apheresis Applications Committee of the American Society for Apheresis are cited. TA has been shown to effectively remove the autoantibodies from blood and lead to rapid improvement.
Keywords
Abbreviations
AA: Aplastic Anemia; AAC: Apheresis Application Committee; AAVs: Adeno-associated viruses; AAV: ANCA associated vasculitis; Ab: Antibody; ABM-ab.GN: Anti-basement membrane antibody; ABO: Blood group system; AChR: Acetylcholine receptor; ADAMTS 1B: A disintegrin and metalloproteinase; ADAMTS13: A disintegrin and metalloproteinase with a thrombospondin type 1 motif. Member13; ADEM: Acute disseminated encephalomyopathy; AID: Autoimmune disease; AIDP:Acuteinflammatorydemyelinatingpolyneuropathy;AIHGA: Autoimmune hemolytic anemia; AKI: Acute kidney injury; ALD: Anti-lymphocyte globulin; ALG: Antilymphpocyte globulin; AMR: Antibody-mediated rejection; ANCA: Antineutrophil cytoplasma antibodies; AntiCD29: Monoclonal antibody; Anti Glu R3: Extracellular antibody; APS: Antiphospholipid syndrome; ASFA: American Society for Apheresis; ATG: Antithymocyte globulin; BANFF: Classification for antibody-mediated rejection; BM: Bethesda Units; BP: Bullous pemphigoid; BW: Body weight; CAPS: Catastrophic antiphospholipid syndrome; CD4, CD25: Regulatory T cells; C5b, C9: Complement components; C3, C3b-C9: Complement factors; CE1, CE3: Cystic echinococconosis; CIC: Circulating immune complex; CIPD: Chronic Inflammatory demyelinating Polyradiculoneuropathy; CMV: Cytomegalo virus; CNS: Central nervous system; Cr: Creatinine; CR1, CR3: Complement receptor; CRISPR/Cas: Clustered regularly interpaced short palindrome repeats; CSF: Cerebrospinal fluid; D: Dalton DC: Dendritic cell; DNA: Deoxyribonucleic acid; Ds: Double strang; DSA: Digital subtraction angiography; ECP: Extracorporeal photopheresis; EHEC: Enterohemorrhagic E. coli; ESRD: End stage renal disease; F: Female; F VIII: Factor VIII; F VIIa: Factor VIIa; FC, FCE I-III: Fragment crystallizable receptor; FcRI-10: FC receptor 10; FFP: Fresh frozen plasma; FSGS: Focal sclerosing glomerulosclerosis; GABHS: Group A beta hemolytic Streptococcus; GBM: Glomerular basement membrane; GBS: Guillain-Barré syndrome; GFR: Glomerular filtration rate; GI:
Gastrointestinal tract; GN: Glomerulonephritis; GNS: N-acetyl glucosamine 6 sulfate; G6PD: Glucose-6-phosphat-dehydrogenase; GP-Lib IIIa, 1b/IV: Glycoprotein IIIa, b/IV; GQ1b: Glycan monoclonal antibody; GVHD: Graft-versus-host-disease (a-; c-); HDN: Hemolytic disease in newborns; Factor HF1: Hageman factor 1; HIV: Human deficiency virus; HLA: Human leukocyte antigen; HMA: Human monoclonal antibody; HPA: Human platelet antigen; HPC: Hematopoietic cell; HPCT: Hematopoietic progenitor cell transplantation; HSCT: Hematopoietic stem- cell transplantation; HSP: Henoch-Schönlein purpura; IA: Immunoadsorption; IC: Immune complex; ICN: Immune complex nephritis; IgA, IgG, IgM: Immunoglobulin IgA, IgG, IgM; H pylori: Helicobacter pylori; IF: Interferon; IL: Interleukin ITP: Idiopathic thrombocytopenic purpura; IUT: Ultrasound tomography; IVIG: Intravenous immunoglobulins; LDH: Lactate dehydrogenase; LDL: Low-density lipoprotein; LEMS: Lambert- Eaton myasthenic syndrome; LRP 4: Low-density lipoprotein receptor related protein 4; M: Male; MCGN: Minimal change glomerulonephritis; MG: Myasthenia gravis; MEPEX: Plasma exchange for renal vasculitis study; MFS: Miller-Fisher syndrome; MiU: Malmö inhibitor units; 8-MOP: 8-Metoxypsoralen; MPA: Microscopic polyangiitis; MMF: Mycophenolate mofetil; MNC: Mononuclear cell; MPGN: Membranoproliferative glomerulonephritis; MRS: Magnet resonance spectroscopy; MRI: Magnetic resonance imaging; MS: Multiple sclerosis; mTOR: Mechanistic target of Rapamycin; Musk: Muscle specific kinase; NC1: No collagenous Domain 1; NMJ: Neuromuscular junction; aRNP: Ribonucleoprotein; NS: Nephrotic syndrome; OKT 3: Monoclonal antibody Orthoclone; PAIg: Plasminogen activator inhibitor; PANDAS: Pediatric autoimmune neuropsychiatric disorders associated with Streptococcal Infections; PEXIVAS: Plasma exchange and Glucocorticoids for Treatment of Anti- Neutrophil Cytoplasma Antibody-Associated Vasculitis; PG: Pyoderma gangrenosum; MPL: Monophosphoryl Lipid A; PRA: Panel-reactive antibody; PRCA: Pure red cell aplasia; PTP: Post transfusion purpura; PV: Pemphigus vulgaris; QA: Quality assurance; RA: Rheumatoid arthritis; RBC: Red blood cell; RD 3: radarman third class; Res: Reticuloendothelial system; RG: recommendation grade; Rh: Rhesus factor; RhD: Rhesus factor D; RNA: Ribonucleic acid; RNP: Ribonucleoprotein; RPGN: Rapid progressive glomerulonephritis; SC: Seydenham´s chorea; SJS: Steven-Johnson syndrome; SLE: Systemic lupus erythematosus; Sm: Smooth muscle; SS: Single stranded; SS-A/Rö: Sjögren syndrome antigen/ribonuleoprotein complex; SS-A/Rö: Sjögren syndrome type B antigen. Anti-1a; TA: Therapeutic apheresis; TAC: Tacrolimus; TEN: Toxic epidermal necrolysis; Th 1 Th 2: T helper cell type 1, type 2; TMA: Thrombotic micro antipathy; TNFα: Tumor necrosis factor α; TPE: Therapeutic plasma exchange; TPO: Thrombopoietin; TPO: Thrombopoietin receptor antagonist; TPV: Total plasma volume; Treg: Regulatory T cell TTP: Thrombotic thrombocytopenic purpura; UVA: Ultraviolet A; WAIHA: Warm autoimmune hemolytic anemia.
Introduction
Therapeutic apheresis is the generic term for different extracorporeal blood purification methods that removes inflammatory mediators, antibodies, and other toxic substances, which are pathogenic in various diseases, from blood. TA is used in many autoimmune diseases [1]. Since the pathogenetic relevance of autoantibodies could defined in various diseases, disease-specific adsorbers have been developed for example, in dilated cardiomyopathy (β1-adrenergic receptors), in systemic lupus erythematosus (SLE, C1q), and in grouping ABO blood group antigens [2-5].
With the hollow fiber modules in TA, a complete separation of the corpuscular components from the plasma is reached and due to increases blood flow rate higher efficacy [6]. TA using centrifuges has shorter treatment times such as TA using hollow fibers shown by Hafer et al. is no advantage [7]. More important is to keep the blood levels with antibodies, and/or pathogenic substances on a very low level over long time during the treatment. However, the substances that should be eliminated could invade into the intravascular space and be eliminated by the membrane separators. Furthermore, cell damage – especially to thrombocytes – occurs less using membranes than centrifuges for all cell separation. The adsorption technologies allow the most selective separation of plasma components without the use of any substitution solution [6]. Membrane techniques are simple and safe to apply and can be competitive to other plasma separation and treatment technologies [8].
The hollow fiber modules in therapeutic plasma exchange (TPE) are mostly used in nephrology, as many of these membranes can be used with currently available dialysis equipment. Nephrologists have an extensive training in the management of blood purification treatments including vascular access, anticoagulation, volume management and prescription for solute clearance [9]. The renal indications for TPE expand the clinical practice of nephrologists [10].
Only a few prospective controlled trials are available that are of adequate statistical power to allow definitive conclusions to be reached regarding the therapeutic value of TPE. This drawback reflects, in part, the relative rarity of most of the disorders under investigation. To compensate, many investigators have understandably grouped heterogenous diseases together, often retrospectively, and used historical controls. The latter design is potentially hazardous, given that earlier diagnosis, recognition of milder cases, and improved general care over time may be lost as a benefit of TPE [11]. Most histories of many diseases commonly treated by TA (e.g., cryoglobulinemia, SLE) are characterized by episodes of exacerbation and remission.
The thresholds for intervention and the details of treatment protocols may vary widely between centers, rendering it difficult to compare studies. TA is primarily used in the treatment of inflammatory renal diseases as an adjunct to conventional immunosuppressive therapy and might be expected a priori to confer only small additional benefit that require large sample size for its detection. Negative studies are inevitably less likely to be published and estimations of efficacy made based on published reports may be based in favor of TA [11].
For the diseases for which the use of TA is discussed, the guidelines on the use of TA from the Apheresis Applications Committee (AAC) of the American Society for Apheresis (ASFA) are cited [12,13]. Bambauer et al. discuss the TA methods such as TPE and different semi- or selective plasma exchange methods [14].
Autoimmune Diseases (AID)
AID caused by antibodies acting against the body´s own tissue. In more than 80% of the illness are common characteristics of which immune mediated destruction of intracellular structures in connective tissue, resulting in fibrinoid tissue damage [2]. AID, with the exception of rheumatoid arthritis and autoimmune thyroiditis are individually rare, but together affect approximately 5 % of the population in western countries: The most of them are poorly understood group of diseases [15]. Viral infections and other influences can lead to altered native antigen with a loss of suppression [2]. Typically, antinuclear antibodies are to be found against most nuclear structures. Vasculitis is common to all diseases, and is most easily demonstrated histological in the precapillary arterioles and post-capillary venuoles: In immune- mediated synovitis, the same chronic cellular infiltrate are seen, with its clinical manifestations of arthralgia or arthritis [16].
The cause of autoimmune reactions is still generally unknown. The spectrum of AID ranges from those diseases in which auto- immunization are solely responsible for the disease condition, to those in which it possibly has a major influence on the further course of the diseases, and those in which the auto-immunization phenomena are probably only of diagnostic importance [16]. These autoantibodies can also be directed at all blood cells.
Inflammation is a complex set of events accompanied by release of much different soluble substance as antibodies (ab) that diffuse away from the site of their production. Autoantibodies are not necessarily auto aggressive or destructive. They only lead to inflammatory tissue reaction when, through their binding to cells and through complement activation, the reaction chain of the serum complement system is triggered [16].
Immune complex (IC) is a physiological process and serves to eliminate foreign material, such as bacteria, their components and viruses. The formed ICs are removed from blood by the adhesion of the Fc-fragments of the antibodies to the corresponding phagocyte receptors in the liver and spleen. ICs are deposited preferentially in certain sites throughout the body, the kidneys, the joints, the lungs and the skin. The kidney accumulates ICs because the blood pressure in the glomerular capillaries is four times higher than in other capillaries and because the glomerulus retains ICs by a simple filtering effect [16]. Similarly, ICs may also accumulate in other body filters. Circulating ICs (CIC) are involved in the regulation of various immune phenomena. With TA, it is possible to interrupt the pathological process by eliminating antibodies.
Nephrological Diseases
Rapid Progressive Glomerulonephritis (RPGN)
RPGN is a diffuse glomerulonephritis that frequently begins acutely. RPGN is a histologic diagnosis, and can occur from a number of etiologies, including anti-basement membrane antibody glomerulonephritis (ABM-ab-GN), which is very rare, antineutrophil cytoplasma antibodies (ANCA), and even IgA nephritis. The histological characteristics are usually capillary emboli with necrosis of the capillary walls and semi-lunar formation, and deposition of IgG and C3 along the glomerular basement membrane. Most cases are simultaneously accompanied by acute kidney injury [17]. More than 90 percent of patients with RPGN due to Goodpasture´s/anti-GBM RPGN have anti-GBM antibodies in their circulation.
RPGN consists of rapid loss of renal function with the histologic finding of crescent formation in over 50% of glomeruli [17]. Histologically is observed a proliferation of cells within Bowman´s space of the glomerulus due to the extravasations of proteins into the space. These cells consist of proliferating parietal epithelial cells as well as infiltrating macrophages and monocytes. RPGN is not a single disease entity but is a clinical syndrome with a different number of etiologies. Linear disposition of IgG due to autoantibodies to type IV collagen representing antiglomerular basement GN (15 %) [17].
The incidence is 0.85 per 100.000/year. Importantly, when discussing RPGN, a number of entities are frequently included in case series and trials, thus confounding results (18). Therapy consists of high-dose corticosteroid (e.g., methylprednisolone) and cytotoxic immunosuppressive drug (e.g., cyclophosphamide or azathioprine) [13]. Other drugs have been used include leflunomide, deoxyspergualin, tumor necrosis factor blockers, calcineurin inhibitors, and antibodies against T cells, or human monoclonal antibodies.
The rationale for therapeutic apheresis is that RPGN with dialysis dependence (Cr > 6 mg/dL) and RPGN with diffuse alveolar hemorrhage have the Category I with the recommendation grade (RG) 1A and 1C. RPGN dialysis independent has the Category III with the RG 2C [12]. Because of the benefit of plasma exchange in the crescentic GN of anti-GBM, TPE was applied to all causes of RPGN [13]. The role of TPE has been examined in some trials in pauci-immune and immune complex GNs and in the treatment of pauci-immune GN. Results of other trials indicate that TPE may be beneficial for dialysis-dependent patients presenting with severe renal dysfunction; however, is no therapeutic benefit over immunosuppression in milder disease. The predominance of pauci-immune GN cases in these series may account for these results [13]. IA is the extracorporeal method that most effectively removes pathogenic immune complexes and antibodies [19]. The frequency of TA is every other day. The volume treated is 1 –1.5 total plasma volume and the substitution solution could be a human-albumin electrolyte solution. Treatment is for 1 – 2 week followed by tapering with less frequent treatments. The duration of therapy is not well defined in the literature. Some trials have stopped TA if there is no response after 4 weeks of therapy. PEXIVAS, an international randomized controlled study comparing TPE versus no TPE and standard versus reduced dose steroid regimen on the primary composite outcome of end stage renal disease (ESRD) or death in patients with ANCA-associated vasculitis (AAV) represents the largest study on the role of TPE in AAV [20]. In the patients under TPE was not significantly associated with their risk of primary outcomes, mortality, and side effects. It was suggested that TPE might be effective in suppressing ESRD in the early stages of treatment [21]. The PEX1VAS study did not show the addition of TPE to standard therapy conferred benefits in patients with severe ANCA-associated vasculitis, but it did show that a reduced-dose regimen of oral glucocorticoids was non-inferior to a standard-dose regimen [22].
Anti-Basement Membrane Antibody Glomerulonephritis (Goodpasture Syndrome, ABM-ab-GN)
In ABM-ab-GN, antibodies appear which that are directed against a peptide component of one of the two non-collagen parts of type IV collagen. However, type IV collagen is found not only in the kidney, but also in the vessels of other organs, such as the lung [23]. The mechanisms responsible for the production of antibodies against the antigens are still not clear.
A large number of diseases have been associated with Goodpasture syndrome based on different cases; however, the most consistently reported associations are with membranous nephropathy and antineutrophil cytoplasmatic-associated vasculitis. Only a small part of ANCA GN have anti-GBM ab, mostly it has thought to be an environmental or infectious exposure that triggers onset of these diseases. It is reasonable to speculate that for both membranous and ANCA-positive vasculitis damage to the kidney elicits an immune response against the GBM, leading to the production of antibodies, which may or may not contribute to disease progression [23]. ANCA GN responds to TPE even when patient on dialysis and anti-GBM GN does not.
The formation of anti-basement membrane antibodies is frequently limited in duration. The autoantibodies cause severe disturbances in the permeability in the lung with significant deterioration in diffusion capacity and hemoptysis. The renal deposition of this autoantibody frequently leads to rapid deterioration in renal functioning, which expresses itself histologically in a necrotizing glomerulonephritis in part. Linear deposits of IgG can be immunohistologically detected both at the basement membrane of the lung, as well as of the kidney [21,22]. An antigen with a probable size of 26,000 – 28,000 daltons is considered responsible for these deposits, its immunogenic epitopes being located on the stable glomerular domain NC1 of collagen IV [22]. The antigen is primarily present in a hexamers form and forms monomers and dimers [22,23]. Antigen determinants are exposed after dissociation and can thus bind specific antibodies. This molecule seems to be present in all basal membranes, in particular in those of the glomeruli, renal tubuli, the Bowman capsule, the lung, and the plexus chorioideus, in the placenta, but also in those of the aorta and the small intestine.
De Lind van Wijngaarden et al. observed that chronic and acute tubulointerstitial lesions predict the glomerular filtration rate (GFR) at 12 months, yet it was the use of TPE and the number of normal glomeruli on biopsy that remained positive predictors of dialysis independence in the same time interval [24]. This is important because it suggests that unaffected glomeruli determine long-term renal outcome at 1 year. In a second study, the same group extended their work in determining the rate of renal recovery [25]. In the MEPEX study, 69 dialysis-dependent patients who were part of the TPE trial, TPE was superior to pulse methylprednisolone with respect to the change of coming of dialysis. The outcome measure depended on the relative number of normal glomeruli.
TA also provides the possibility of improvement in cases of pulmonary bleeding, which based on the same immunological process, even when renal function is already irreversibly impaired [26,27]. A final long-term prognosis for patients whose condition improved after TA cannot be made. As basement membrane antibody formation often ceases during treatment, recovery, or at least partial recovery is possible.
The rationale for TA is that RPGN with dialysis dependence (Cr > 6 mg/dL) and RPGN with diffuse alveolar hemorrhage have the category I with the RG 1A and 1C. RPGN dialysis independent has the Category III with the RG 2C (Table 1) [12]. Because of the benefit of TPE in the crescentic GN of anti-GBM, TPE was applied to all causes of RPGN. The frequency of TA is every or every other day until anti-glomerular basement membrane antibodies are detectable. The volume treated is 1-1.5 total plasma volume, and the substitution solution could be a 5% human albumin-electrolyte solution or fresh frozen plasma (FFP). Treatment is for 1-2 week followed by tapering with less frequent treatments. The duration of therapy is not well defined in the literature. Some trial have stopped TA if there is no response after 4 weeks of therapy. TA should be continued until antibodies fall to undetectable levels in patients with active disease and anti-GN M antibodies present [12].
Immune Complex Nephritis (ICN)
Many types of glomerulonephritis are initiated by the deposition of immune complexes (ICs), which induce tissue injury via either engagement of Fc-receptors on effector cells or via complement activation [28]. The pathogenic consequences of systemic autoimmune disease is thought to trigger by the generation of antibody and subsequent tissue deposition of ICs. Modulation of the autoantibody response disrupts pathogenesis by preventing the formation of ICs; however, uncoupling IC formation from subsequent inflammatory response seems unlikely because of the apparent complexity of the IC-triggered inflammatory cascade [29].
In idiopathic symptomatic RPGN, which is frequently caused by an immune complex nephritis, the therapeutic concept is not as clear-cut as with anti-glomerular basement membrane antibody.
Table 1: Guidelines on the use of TA in clinical practice-based approach in nephrology diseases [12,13], and own observations from 1980-2008.
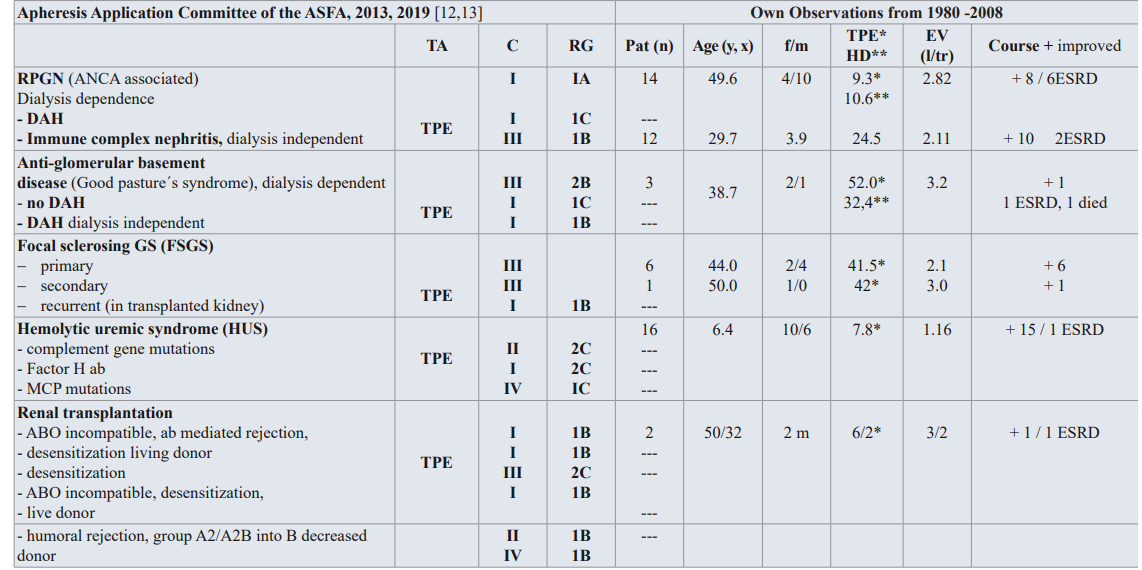
TA: TA modality; C: Category; RG: Recommendation grade; Pat: patient; f/m: female/male; EV: Exchange volume (l/tr: liter / treatment,x);Substitution: 3-5% human albumin electrolyte solution, FFP; DAH: diffuse alveolar hemorrhage, MCP: membrane cofactor protein
Category I: accepted for TA as first-line therapy; Category II: accepted for TA as second-line therapy; Category III: not accepted for TA, decision should be individualized; Category IV: not accepted for TA, approval is desirable if TA is undertaken [12,13].
Nephritis. An improvement in renal function is possible in more than 60 percent of cases, if either pulse therapy (high dose therapy with corticosteroids) or TPE is administered. In view of the devastating pathophysiologic consequences of interaction between circulation immune complexes and the basement membrane was found, that TPE in combination with immunosuppression should be carried out as quickly as possible [30].
Combination of corticosteroid and cyclophosphamide or rituximab, and/or mycophenolate mofetil (MMF), and tacrolimus (TAC) has been recommended for remission induction of ANCA- associated vasculitis [31]. This is the first report demonstrating the efficacy of a multi-target therapy of corticosteroid, MMF, and TAC for remission-induction of intractable ANCA-associated glomerulonephritis developed independently of systemic lupus erythematosus.
RPGN with or without Glomerular Deposition (ANCA ab) Pauci – Immune RPGN
Approximately 60 percent of patients with RPGN present with crescentic glomerulonephritis characterized by few or absent immune deposits, the so-called pauci-immune RPGN. Patients with this disease have either Wegner´s granulomatosis; ANCA- ab associated vasculitis, polyarthritis nodosa, or “renal-limited” pauci-immune GN (Table 1) [32]. These diagnoses may represent a spectrum of manifestations of a single disease, because there is marked overlap of clinical and histopathologic features, and several patients have anti-neutrophil cytoplasmatic antibodies in their blood which are more common that anti-GBM. The concentration of circulating ANCA correlate with the disease activity in some patients, and ANCA may contribute to the pathophysiology of pauci immune RPGN through reactivity with neutrophils or endothelial cells, and other inflammatory mechanisms [32,33].
The prognosis of pauci-immune RPGN in general has been poor. Precise therapy therapeutic recommendations are difficult to obtain from the literature, because most series comprise patients with different types of RPGN. However, available data suggest that 80 percent of such patients’ progress to ESRD without therapy with high dose immunosuppression or cytotoxic drugs. Some trials have evaluated the efficacy of TA as an adjunct to conventional immunosuppressive in patients with pauci-immune RPGN [32,33].
In milder forms of pauci–immune RPGN, the generation of antibody and subsequent tissue deposition of immune complexes, the results of the randomized trials argue against the role for TA; however, suggest a potential benefit when TA is used as an adjunct to conventional immunosuppressive therapy in patients with severe disease. This relative lack of efficacy probably reflects the efficiency of conventional immunosuppressive agents in halting inflammation and preserving renal function in most patients. These conclusions are supported by the results of uncontrolled trials, suggesting a response rate of 70 percent in patients with RPGN treated with TA, similar to that of patients treated with immunosuppressive therapy with a response rate of 60 percent. In most cases of RPGN, a treatment of TA in the early phase of the disease seems to be necessary.
In the MEPEX study, de Lind van Wijngaarden et al. showed that in patients with dialysis-dependent, ANCA-associated vasculitis, the chances of recovery differ depending on the type of adjunctive treatment, the percentage of normal glomeruli and glomerulosclerosis, the extent of tubular atrophy, and the presence of arteriosclerosis. Even with an ominous biopsy at diagnosis in combination – with dialysis dependence, the chance of renal recovery exceeds the chance of therapy-related death when the patient is treated with TPE as adjunctive therapy [25]. The PEXIVAS trial did not show that the addition of TPE to standard therapy conferred benefits in patients with severe ANCA- associated vasculitis, but it did show a reduced-dose regimen of oral glucocorticoids was non-inferior to standard-dose regimen [24]. With high titers of circulating ICs or other antibodies, which could damage the kidney and other organs, IA with protein-A, or sheep polyclonal antibodies can be more effective than the TPE procedure.
Therapy Recommendations for RPGN
RPGN therapy possibilities were extended in recent years to include TA. Antigens, antigen-antibody complexes, and immune complexes can be eliminated from the blood with the aid of TPE. A corresponding therapy enables immunomodulation through suppression or stimulation of antibody formation, as well as a temporary remission of the inflammation through inhibition of the mediators. TPE combined with immunosuppression therapy seems to us to be advisable, particularly in view of the unfavorable prognosis for RPGN, with its complex causes.
The therapy recommendation is based on the few uncontrolled and controlled studies available [32,34-37] TPE is indicated in combination with an immunosuppressive therapy with prednisolone (intravenous pulse therapy, or oral therapy), cyclophosphamide (intravenous pulse therapy or oral therapy), or azathioprine in the following cases:
- RPGN with serum creatinine under 5.8mg/dl without oliguria in anti-GBM disease
- All severe forms of RPGN with or without ANCA ab, like the pauci-immune complexes, (Cr > 6 or patient on dialysis)
- Goodpasture syndrome with life-threatening hemoptysis, or diffuse alveolar hemorrhage from ANCA or MPA independent of renal function status
TA used for renal indications, even in elderly patients is relatively safe. Trends towards death in elderly patients may be multi- factorials and not necessary related to TA (37). TA may be decrease end of end-stage renal disease or death in patients with RPGN [38]. The combination of TA with immunosuppressive therapies including biologics seems to be more effective as TA alone, but additional trials are required. However, other authors prefer in cyclophosphamide-resistant ANCA-associated GN a multi-target therapy, a combination of corticosteroids, MMF, and TAC; however, additional trials are required [31].
Glomerulonephritis with Nephrotic Syndrome (NS) Classification is classified morphologically, and thus does not provide a uniform description of the disease. Differing etiologies can result in considerable variations in the clinical features, as well as course and prognosis. Therefore, it is difficult to establish generally applicable therapeutic concepts and customized treatment for the individual patient is the norm [39]. The variable clinical courses of this heterogenous disease group render it almost impossible to carry out controlled therapy studies. The clinical successes and failures are to be found, as are therapy-produced complications, e.g., infections, sterility, loss of hair, and others. The benefits of immunosuppressive therapy must be weighed against these complications. The aim of therapy for glomerulonephritis is to prevent terminal renal insufficiency and the risks of nephrotic syndrome.
The cause of NS lies in changes in the electrophysiological characteristics of the filtration barriers and of the plasma proteins. The anionic charge on albumin is retained by the negative charge of the glomerular filter - including the basement membrane and the epithelium - obviously play a decisive role [11]. Hemodynamic changes, such as increase in venous pressure, can favor the filtration of proteins.
NS of various GN often reacts to corticosteroids in varying doses, administered over a period of 4 - 8 weeks. Patients with frequent relapses are also treated with 2 - 3 mg/kg BW/day cyclophosphamide [17]. Cyclosporin A has also been successfully applied in nephrotic syndrome [6]. High doses of immunoglobulin (IgG) for nephrotic syndrome, administered 0.4 g/kg BW IgG on three successive days were reported and repeated every 21 days over a period of one year. Other therapeutic measures for nephrotic syndrome are anticoagulants, thrombocyte inhibitors, ACE inhibitors, immunosuppressive drugs, lipid reducers, biologics, and diets [40-42].
The prognosis for focal sclerosing glomerulosclerosis (FSGS), usually accompanied by nephrotic syndrome, is considerably less favorable. Cases with nephrotic syndrome are recorded as having a survival rate of 70 percent after six years. Without nephrotic syndrome, this rate reaches 85 percent. Patients with this form of glomerulonephritis are comprised of steroid – sensitive and a steroid - non-sensitive groups and an appropriate therapy must be selected. Non-reaction to steroids is an indication for a therapy with cyclophosphamide, chlorambucil, or cyclosporin or other immunosuppressive therapy [43]. FSGS is caused by a variety of factors, however, one type that recurs after transplantation and has been associated with circulating factors, can be treated with TPE.
In resistance to medication or severe progression of the disease, additional TA therapy should be considered, as a continuing treatment given once a week, or every two weeks, or once a month. After transplantation, as many as 40 percent of patients with nephrotic syndrome have recurrences. The glomerular abnormalities in patients with established disease include focal and segmental glomerulosclerosis and hyalinosis, although fusion of epithelial-cell foot processes may be the only abnormality early in the course of disease [12]. Some patients with recurrent focal glomerulosclerosis have a response to treatment with TPE, LDL apheresis and IA there may be different circulating factors that alter the glomerular barrier to protein filtration [44].
In the guidelines on the use of TA in clinical practice-evidence- based approach from the AAC of the ASFA has the primary and secondary FSGS the Category III with the RG 1C, and for the FSGS recurrent the category I with the recommendation grade 1B (Table 1) [12,13].
The treatment in native kidneys with FSGS is primarily with corticosteroids for at least 6 months prior to trying second-line agents such as cyclophosphamide, chlorambucil, or azathioprine. For resistant cases, TPE is being currently an option. Investigators worldwide have used TPE in the management of patients with FSGS in transplanted organs, in an attempt to save the graft. There is no standardized treatment for recurrent FSGS post-transplant; the majority of regimens use a combination of an immunosuppressant such as cyclophosphamide, biologics, and TPE. Other therapeutic options include high-dose cyclosporine, angiotensin converting enzyme inhibitors, and indomethacin and/or tacrolimus. Another approach to prevent recurrent FSGS is several sessions of pre- emptive TPE immediately prior to and following the transplant [11]. More recently, rituximab and mycophenolate mofetil have also been used in conjunction with diagnosed in order to halt the process and maintain renal function [12,13].
In certain FSGS patients appears to contain an ill defined “permeability factor”, probably a glycoprotein of molecular weight of 30 – 50 kD that includes profound leakage of albumin when incubated with isolated rat glomeruli. Such factor is removed by TPE and the decrease in serum concentration coincides with improvement in proteinuria. The immediate onset of proteinuria following transplant is mediated by this factor, prophylactic TPE may be instituted in high-risk patients. Some reports describe the use of Staphylococcal protein-A columns in recurrent FSGS. The duration of the procedure is to begin with three daily exchanges followed by at least six more TPE in the subsequent 2 weeks, for minimum of nine procedures. Tapering should be decided on a case- by-case basis and is guided by the degree of proteinuria. Timing of clinical response is quite variable and control of proteinuria may take several weeks to months. Some patients have received long- term monthly exchanges as maintenance therapy [12,13].
The nephrotic syndrome consisting of massive proteinuria, hypoalbuminemia, edema, and hyperlipidemia, is a common complication of glomerular disease in children and adults. The annual incidence of nephrotic syndrome ranges from 2 – 7 per 100,000 children, and prevalence from 12 – 16 per 100,000. There is epidemiological evidence of a higher incidence of NS in children aged below 10 years from South ASIA [41]. The primary cause of NS is idiopathic. There is evidence pointing to a role of the immune system in pediatric minimal change glomerulonephritis (MCGN). Another hypothesis has described an association between allergy and MCGN in children. Relapses in this of syndrome are triggered commonly by minor infections and occasionally by reactions to be stings or poisoning. Abnormalities of both humoral and cellular immunity have been described. Finally, the induction of remissions by corticosteroid, alkylating agents, or cyclosporine therapy provides indirect evidence for an immune etiology [12].
Although they are massively proteinuria, patients with MCGN, do not have a generalized glomerular leak to macromolecules. The clearance of neutral macromolecules in MCGN is actually less than normal over a range of molecular radii. In contrast, the clearance of anionic macromolecules is significantly increased. This and several other lines of evidence suggest that proteinuria results from a loss results from a loss of fixed negative charges of anionic glycosaminoglycan’s in the glomerular capillary wall [12]. The mechanisms through which these charges are lost are unknown. The traditional view is that massive albuminuria, in NS causes a decrease in intravascular oncotic pressure, which allows extravasation of fluid and hypovolemia, increased aldosterone and antidiuretic hormone secretion, and renal salt and water retention. MCGN usually takes a benign course and can be well treated with customary therapy measures. In severe cases, therapy with prednisolone and cyclophosphamide over a period of 8 to 12 weeks is indicated [45,46]. Cyclosporin has shown some efficacy in steroid-resistant NS [47]. A significantly rapid faster relief from steroid–resistant NS by using LDL apheresis than from steroid monotherapy is reported [47]. A rapid improvement of hypercholesterolemia by LDL apheresis in steroid–resistant NS will provide more rapid relief from NS than from steroid therapy alone. Others recommended in steroid-resistant NS intravenous steroids in high dose with alkylating agents, cyclophosphamide oral or pulse cyclophosphamide and mycophenolate mofetil [48].
Membranoproliferative glomerulonephritis (MPGN) usually occurs in combination with nephrotic syndrome and hypertension. The occurrence of NS signifies a poorer prognosis. The effectiveness of medication with corticosteroids or pulse therapy, cyclophosphamides, anticoagulants, and intravenous immunoglobulins has not yet been established [49]. A successful treatment with protein-A IA in patients with relapsing nephrotic syndrome was reported [49,50]. MPGN from cryoglobulinemia could be an indication for TA, too.
NS is the main symptom in peri-membranous glomerulonephritis. In the case of acute nephrotic syndrome, it is advisable to undertake therapy with high doses of prednisolone as a pulse therapy over a period of 3 to 5 days or with 2 mg/kg BW in decreasing dosage for 2 to 3 months. A combination with TPE should be considered especially with the more selective procedures like cascade filtration, IA, and LDL apheresis [47,49,51].
The symptoms in mesangioproliferative glomerulonephritis are not usually homogeneous. The prognosis is poorer if NS and hypertension accompany the condition. There are varying opinions exist with regard to corticosteroid and cytostatic therapy. NS justifies a trial therapy with cyclophosphamide. In severe, drug therapy-resistant cases, a combined TA and immunosuppression therapy is recommended, regardless of the degree of renal insufficiency [52].
Acute NS in particular seems to be favorably influenced by regular TA treatment, for first, dysproteinemias and thus the edema can be improved, and second human albumin can be administered in larger doses. TA is theoretically a way of achieving an improved effect on the basal membrane. The elimination of cholesterol, LDL, and triglycerides might also reduce the atherogenic risk for these patients and thus prevent progression. TA should be considered as a useful therapeutic tool in the management of this disease [44]. The reports of the therapy of NS with more selective TA procedures like cascade filtration, IA, and LDL-apheresis are very encouraging and show a possibility for treating severe cases of NS, if drug therapy fails. Renal diseases such as light chain nephropathy, dense deposit diseases and others can be in severe cases and if the conservative therapy has failed, threated with TPE. As in the case of other renal diseases, controlled prospective studies are needed.
Hemolytic-Uremic Syndrome (HUS)
HUS is a disease that can lead to acute kidney injury (AKI) and often to other serious sequelae, including death. The disease is characterized by microangiopathic hemolytic anemia, thrombocytopenia and AKI. The etiology and pathogenesis of HUS are not completely understood, and the therapy of HUS is complicated. After introduction of therapeutic apheresis as a supportive therapy in HUS, several authors reported successful treatment using TA in HUS in more than 87 percent of treated patients. The supportive therapy is indicated in severe courses of HUS and is superior to available therapy interventions. Bambauer et al. [53] discuss the pathophysiologic aspects of the different pathogenic types of HUS.
Most cases are associated with infections with enterohemorrhagic coli (EHEC). These bacteria can be transmitted through contaminated food, animal and person to person contact. HUS is one of the most severe complications of a potentially avoidable food-borne infection. Other causes of HUS described as “typical” have to be differentiated since other factors including genetic disorders are of importance. A minimum of three different pathogenetic types, which lead to HUS, are subdivided. HUS caused by infection, idiopathic HUS (non-Shiga toxin HUS), and HUS in systemic diseases and after toxin exposure [54].
There have been reports of spontaneous recovery from HUS. The various etiological and pathogenetic assumptions have produced diverse therapy concepts. However, the total lethality in HUS was first reduced to 20 percent with the introduction of dialysis [55].
If the therapy is administered early enough, two-thirds of cases recover without any impairment. In 10 - 20 percent of cases, however, lasting renal damage occurs. Other authors reported successful in HUS using TPE and successful treatment in HUS using IA with protein-A [56-59]. A compilation of therapeutic concepts for HUS implemented up to 2009 showed the success of HUS therapy with TPE/HD or IA/HD [53].
Substitution of plasma or coagulation factors is often necessary due to the severe coagulation problems in HUS. TA might be more effective than infusions alone, as it removes potentially toxic substances from the circulation. TPE or IA should be considered first-line therapy in situations that limit the amount of plasma that can be infused, such as renal or heart failure. Plasma infusion treatment is contraindicated in S. pneumonia induced non-Stx- HUS. It may exacerbate the disease because adult plasma contains antibodies against Thomson Friedenreich antigen [60].
Different randomized controlled trials showed that TPE and/or dialysis as supportive therapy are still the most effective treatments in HUS [61]. The outcome was listed for HUS, all-cause mortality, chronic reduced kidney function, and persistent proteinuria or hypertension at last follow up. None of the evaluated interventions such as fresh frozen plasma transfusion or dipyridamole, Shiga toxin binding protein and steroids was superior to supportive therapy alone for any outcomes [61].
The advantage of TA over other therapeutic procedures is that it intervenes at an early stage in the pathogenetic processes by quickly removing immune complexes and toxins. TA eliminates fibrinogen, fibrinogen degradation products, and other high molecular complexes, all of which can both support and inhibit coagulation. All other toxins produced by bacteriae and viruses like Shiga-toxin, the pathogenic pathway which follows the activation of the complement system of the factor HF1 with a partial HF1 deficiency and all other toxic substances can be quickly removed by TA.
TA methods, which are introduced in HUS as a supportive therapy, are TPE and IA with protein-A columns. Both methods are described elsewhere [53,56,59]. The rationale for TA in HUS is discussed controversially because of the limited and/or conflicting data available in the literature. The rationale is that TA can effectively remove antibody or mutated circulating complements regulators [12]. TA seems a reasonable option considering the poor prognosis of HUS in adults [53]. The role of TA is uncertain but this treatment may be appropriate as supportive therapy.
The AAC of the ASFA divided HUS in 3 groups for TPE: Group 1 (diarrhea associated HUS) is a HUS due to complement factor gene mutations has the category II with the RG 2C. Group 2 is a HUS due to autoantibody to factor H (atypical HUS), and has the category I with the RG 2C. Group 3 is the typical HUS < 18 years. Group 3 has the category IV with the RG 1C (Table 1) [12,13]. Due to the various and very different causes, which can lead to a hemolytic-uremic syndrome, there are no exact guidelines available for the therapy of HUS.
In HUS, a supportive therapy is indicated which include control of fluid and electrolyte imbalance, use of dialysis if required, control of hypertension, blood and plasma transfusion as required. Antibiotic treatment of E. coli O157:H7 colitis may stimulate further verotoxin production and thereby increase the risk of HUS. The use of dialysis like hemodialysis or peritoneal dialysis as required daily. However, untreated HUS in adults and children may progress to end in organ damage [62]. Platelet transfusion may actually worsen outcome.
TPE or IA is generally performed daily until the platelet count is normal. In TPE, the replacement fluid consists of human albumin- electrolyte solution (5%) in 30 to 70 percent and FFP in the remainder. The exchange volume per treatment should be 1 – 1.5 total plasma volume (TPV) depending on the severity of the HUS. TPE may reverse the ongoing platelet consumption. By using IA, no replacement fluid is necessary only between the treatments FFP or coagulation factors may be transfused if required. The hemodialysis treatment can be combined with the TA.
A large outbreak of diarrhea and the HUS caused by an unusual serotype of Shiga-toxin-producing Escherichia coli (O104:H4) was in Germany in May to July 2011 with 3,167 without HUS and 16 deaths in the patients, and 908 with HUS and 34 deaths [63]. 241 patients with HUS were treated with TPE and 193 patients with TPE and eculizumab. The treatment strategy was dependent on disease severity [64]. TPE and eculizumab in combination seems to be prudent and necessary prior to establishing new treatment guidelines.
Kidney Transplant Rejection
In chronic renal failure, a kidney transplantation is the decisive alternative to permanent dialysis. Rejection of the transplanted kidney is a grave problem. Although various therapeutic interventions to delay or prevent rejection exist and use steroids, immunoglobulins, immunosuppressives, cyclosporine A, triple drug, OKT3, and other new developed immunosuppressive therapies. Infections and rejection reactions are the most frequent complications of modern transplantation [65,66]. Thus, acute kidney transplant rejection is considered as an indication for TPE [66,67]. TA is indicated in the management of rejection crisis due to preformed specific antibodies or a high degree of immunization [65].
Immunological problems like performed donor-specific antibodies or a high degree of immunization complicate the outcome of donor transplantation. Postoperatively the antibody-mediated rejection or drug-related side effects of the medication can limit the therapeutic success of transplantation. Acute allograft rejection is one of the important complications after renal transplantation, and it is a deleterious factor for long-term graft survival. Rejection is a complex pathophysiologic process, which has been explained by transcriptome and proteome in RNA transcripts and proteins level respectively [68,69]. Therefore, therapeutic strategies include a primary avoidance of immunization, careful patient selection, a meticulous immunological workup and a proper follow up and TA as improved therapy [70,71].
After the blood group barrier had been successfully crossed in Japan in the 1980s, different protocols were developed for ABO-incompatible kidney transplantation and the procedure has gained widespread acceptance and has been implemented in most transplant centers [68,69]. Immunosuppression consists of tacrolimus, mycophenolate and steroids together with induction therapy with an IL-2-receptor blocking agent. The isoagglutinine antibodies against the donor can be eliminated. Firstly, the CD 19/20-positive pre-B cells with a single infusion of rituximab four weeks prior to transplantation and in a second step, the already existing antibodies are depleted by using TA such as TPE or IA. Novel sensitization and production of antibodies is thereby efficiently prevented [71,72].
The disadvantage by using TPE is the elimination of physiological proteins, the limitation to 1 – 1.5 TPV as treating dose and the potential for infectious complications such as HIV or hepatitis B or C by using plasma as substitution solution. Therefore, various groups use the IA with unselective IgG columns. Patients with performed HLA antibodies, i.e. a high percentage of panel reactive antibodies, accumulate on the waiting list for kidney transplantation and can experience a substantially longer waiting time [65,71]. Therefore, center specific desensitization protocols were developed in order to transplant these highly immunized patients within a reasonable period.
The transplantation procedure is problematic with deceased donor organs as the time for pre-conditioning of the recipient is extremely limited and the accompanying procedures are difficult to perform in time. If transplantation from a living donor with DSA is planned, different protocols were published to desensitize the recipient. These strategies require an intensive procedure, mostly consisting of the administration of intravenous immunoglobulins (IVIG), of intensified immunosuppression, pre- and postoperative TPE or IA and carry a higher risk for antibody-mediated rejection [65,73-75]. TA in all forms can be applied to remove DSA and multiple HLA antibodies. No selective secondary adsorbers exist, and available columns with a selectivity for immunoglobulins would be considered the best option. Some treatments are usually needed to deplete to recipient of the DSA- and/or anti-HLA titer.
Acute antibody rejection of organ allografts usually presents as severe dysfunction with a high risk of allografts loss. HLA antibodies are involved in AMR [76]. The renal biopsy often cannot rule out one cause or the other with sufficient certainty, leaving the physician with the decision how to treat vascular rejection that can be caused by antibodies or cellular infiltration [77]. TA accompanied by T cell depletion (ATG, ALG, or OKT3) conversion to a tacrolimus-based immunosuppression and pulsed steroids, are used to limit the interstitial and vascular damage [75].
The use of IA targeted against IgG has been used successfully. It is not possible, due to conflicting and limited data, to give general recommendations about the treatment of TPE or IA, the number of apheresis sessions and the best immunosuppressive therapy [78]. A screening for donor-specific antibodies should be performed to monitor the antibody titer during treatment, until 10 sessions with daily treatments initially followed by apheresis every other day can be necessary in a patient with vascular rejection (Banff IIb-III or AMR) [65,74].
Recurrence or de novo thrombotic microangiopathy (TMA) in the transient patient is observed rarely with the use of calcineurin inhibitors or mTOR inhibitors or acute vascular rejection. Infectious diseases such as HIV, CMV, Para virus B 19, an inhibited or decreased activity of the von Willebrand factor- cleaving metalloprotease ADAMTS13 or mutations in complement receptors may also trigger microangiopathy with either limited or systemic manifestations [65].
TA can be attempted to ameliorate the course of the disease and subsequent graft damage, if switching to a different immunosuppressive regimen or the treatment of an underlying infection does not lead to an improvement of the TMA [78]. The treated volume is usually one TPV with human albumin and/ or fresh frozen plasma as substitution fluid and anticoagulation with heparin on a daily basis until platelet count and lactate dehydrogenase have normalized. Up to 50 percent of patients demonstrate a prompt exacerbation if daily TA is stopped. Continuation of TA on an alternate day strategy for at least two additional treatments can reduce the recurrence rate. Nevertheless, TMA reduces graft survival both in recurring or de novo TMA and treatment might not alter the progression of the disease [65].
Goodpasture syndrome or anti-GBM disease can occur de novo in patients following transplantation or as a manifestation of underlying Alport disease, but is rare (e.g., 3 percent of transplanted male Alport patients) [79-81]. The recipient´s immune system is exposed to a collagen component carried by the transplanted organ that is lacking in Alport patients and, consequently, the patient might develop antibodies against this antigen in the glomerular basement membrane. These antibodies may then induce post- transplantation anti-GBM disease.
The treatment of this condition and of de novo disease is identical to the strategy applied to non-transplanted patients. Both TPE and IA have been shown to deplete the patient effectively of antibodies and halt disease progression [82,83]. The TA should be a rapid removal of the antibodies with daily treatments. Treatment frequency should be tapered later to antibody titer measurements. TA is accompanied by an intensified immunosuppressive regimen to suppress further antibody formation [65,84].
Only some information is available about long-term results of kidney transplantation in adults with focal segmental glomerulosclerosis. However, primary FSGS recurs with uncertain incidence after kidney transplantation (presumably 20 percent). A circulating factor is assumed to play a causative role and TA has been successfully applied in patients with recurrent FSGS. In patients treated with TPE, or with a protein-A adsorption column, a dramatic but usually transient reduction in proteinuria has been described [85]. This effect was larger with the use of IA, but remissions that are more prolonged were reported with the use of TPE with or without combination with cyclophosphamide [65,84].
TA in transplantation as an important part of different therapy strategies like for therapy of several conditions such as AMR or ABOi transplantation is accepted today. TA enables the physicians to develop strategies to provide the best organ replacement to patients with high degree of immunization or performed DSA thereby expanding the use of living donation. The standard method has been TPE but it is currently more and more replaced by the more selective methods provided by immunoadsorption. Due to the considerable costs of IA, the selection and application of an adsorber and device for IA should be preceded by a judicious effort to characterize and plan the treatment.
The guidelines of the AAC of the ASFA describe the antibody- mediated rejection and HLA desensitization as follows and give for the AMR renal transplant recipients and desensitization living donor due to donor specific HLA antibody the category I with the RG 1B. The desensitization high PRA deceased donor has the category III with the RG 2C [12,13].
AMR affects less than 10 percent of renal allografts. Recipients at increased risk include those with previous transplant and high panel reactive antibodies [12]. New immunosuppressive drugs are continually being developed to prevent and treat acute allograft rejection. All transplant recipients are placed on immunosuppressive therapy but individuals with a high likelihood of acute rejection, including those with HLA antibodies and recipients of cadaveric organs, receive regimens that are more intensive. The optimal regimen has yet not to be defined but include the use of cyclosporine, tacrolimus, mycophenolate mofetil, azathioprine, and ant lymphocyte globulin [17,18]. Other monoclonal antibodies are rituximab, bortezomib and eculizumab [12,13].
The rationale for TA is that AMR and DSA, which are generated after transplantation, can be removed with TPE, double filtration plasmapheresis, lymphoplasmapheresis, and IA [11]. TPE is used to lower antibody titer below a critical threshold. TPE has been included in preparatory regimes for ABOi renal transplantation in addition to other immunosuppressive/immunomodulatory drugs therapies; this is likely due to improved anti-rejections, improved detection of DSA, and improved definition of AMR using the Banff criteria. Previously there was a high graft loss rate with acute vascular rejection, current regimens, which include TPE, have a graft survival rate of 70 – 80 percent [11].
TA can also be used prior to transplant to remove HLA antibodies. TPE is used in combination with immunosuppressive drugs pre- transplant until cross-match is negative. TPE is usually continued post-operatively and reinitiated in cases where AMR occurs. The ability to obtain a negative cross-match depends on the DSA titer. Using approximately five TPE preoperatively, will allow the titer of ≤ 32 to become negative. The risk of AMR is approximately 30 percent with a small number of graft losses. The desensitization protocols should be used only in highly selected patients [18].
Patients should be started on immunosuppressive drugs prior to initiate TPE to limit antibody re-synthesis. For desensitization protocols, there appears to be a correlation between the number of TPE needed pre-operatively to obtain a negative cross-match and the antibody titer [12]. The exchange volume will be 1 –1.5 TPV and the replacement fluid can be a human-albumin (5 percent) electrolyte solution or FFP. TPE is also performed post- operatively for a minimum of three procedures. Further treatment is determined by risk of AMR, DSA titers, or the occurrence of AMR [13,86].
Further investigations and more controlled studies will show the importance of TA in the therapy strategies, but the financial aspects of TA are matter of regional negotiation and preference. To simplify reimbursement, transplant centers should define their needs aim for a standard reimbursement and to try to limit price variations of this very expensive therapy [65].
Neurologic Disorders
Neurological disorders constitute the largest group of indications for TA [87]. Severe central nervous system (CNS) involvement is associated with poor prognosis, and high mortality rate. High dose steroid and cyclophosphamide (oral or intravenous) are the first choice of drugs in the treatment; TA, intra-venous IVIG, thalidomide, intratechal treatment may be valuable in treatment resistant and serious cases. Table 2 shows the most of the neurological diseases that have been treated with TPE with the categories and the RG of the AAC [12,13].
Acute Inflammatory Demyelinating Polyneuropathy (AIDP)
(Guillain-Barré Syndrome (GBS)
AIDP is an auto aggressive disorder that develops subsequent to infectious diseases and because of other noxae [17]. It is an acute polyradiculitis, which mostly affects the distal and proximal muscles of the extremities, as well as the trunk muscles and can progress with severe ascending paralysis, ending in respiratory paralysis [88,89]. Most patients with AIDP have inflammatory, predominantly demyelinating polyneuropathy. This acute progressive disease, leading to rising paralysis, usually reaches its height within one to two weeks; 25 percent of all patients require artificial ventilation. AIDP occurs in one out of 50,000 persons each year in the industrial nations, regardless of gender or age [17].
The pathophysiologic mechanism has not been established completely, but in many cases, an antecedent infection by campylobacter jejuni leads to the production of antibodies (abs) directed against certain epitopes of the bacterium that also destroy the myelin sheath of the peripheral nerve. This phenomenon has been described as molecular mimicry [90]. The spectrum of organisms responsible for infections can trigger GBS ranges from Eppstein-Barr virus to mycoplasma, herpes zoster, and mumps virus, borrelia to the HIV viruses [91]. However, AIDP directly attacks the myelin sheath, resulting in segmental demyelination and remyelination.
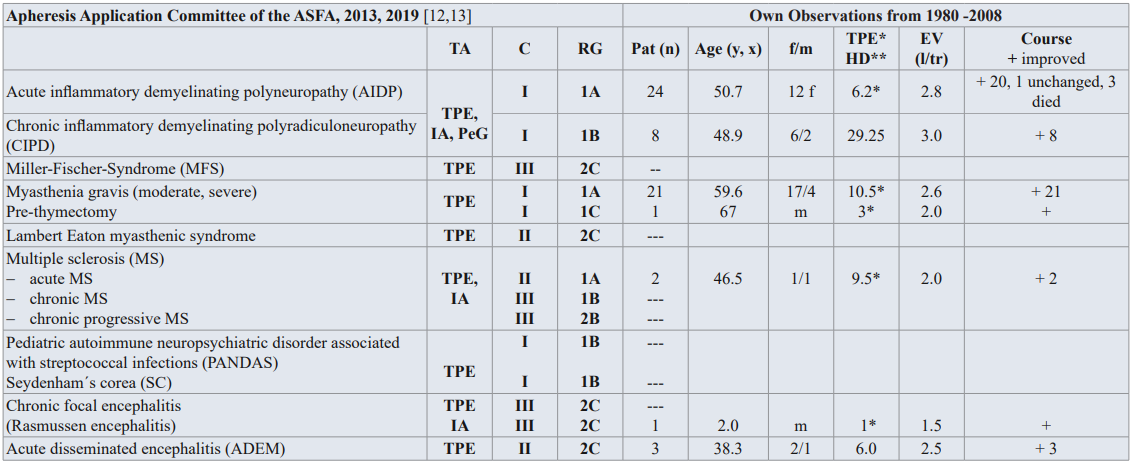
Table 2: Guidelines on the use of TA in clinical practice-based approach in neurology diseases [12,13], and own observation from 1980-2008.
TA: TA modality; C: Category; RG: Recommendation grade; Pat: patient; f/m: female/male; EV: Exchange volume (l/tr: liter / treatment,x); Substitution: 3-5 % human albumin electrolyte solution, FFP; PeG: Peptid-Gam®: synthetic peptide-goat-antimouse; IA: Immonadsorption on protein A
Category I: accepted for TA as first-line therapy; Category II: accepted for TA as second-line therapy; Category III: not accepted for TA, decision should be individualized; Category IV: not accepted for TA, approval is desirable if TA is undertaken [12,13].
In recent years, the triggering causes have been described as being: 1) Antibodies against peripheral nerves, in particular against myelin; 2) circulating immune complexes; 3) complement activation in the cerebrospinal fluid and in serum; 4) other inflammatory mediators and cytokines; and 5) a disorder in cell- related immunity [17].
Electro-diagnostic study is the accepted standard for differentiating between axonal and myelin lesions in early-stage acute polyneuropathy. However, current electro-diagnostic criteria have some limitation in diagnosing axonal GBS [92]. The axonal type of GBS is pathophysiologically characterized not only by axonal degeneration, but also by reversible conduction failure. Antiganglioside antibody tests will facilitate a correct diagnosis. However, there are seronegative AIDP patients, too [93].
Spontaneous recovery normally occurs between the 2nd and 4th week of illness, and, in 75 percent of the patients, it can even occur after several months of illness. Due to remaining damage and relapses, lethality is between 5 and 25 percent after one year. The rationale for TA is based on the humeral and cellular immune dysfunction in this disease [94].
Intra-venous immunoglobulin has also been shown to be effective in the treatment of AIDP. In a recent large international randomized study of TPE, IVIG, and combined treatments in AIDP, all three modalities were effective [95]. TPE was noted to be better than IVIG, and the combination was better than either of the treatments alone [96,97].
In recent years, researchers have applied a combination therapy of TPE or IA following by IgG (0.4g/kg BW for 5 days) [94]. Haupt et al. reported results which suggesting that such a combination treatment of AIDP may be superior to plasma exchange alone [98]. Accordingly, with TPE treatment in GBS, it was possible to reduce the costs by between 30 to 40 percent in America, due to the shorter periods of inpatient treatment and shorter duration of artificial respiration [17,99]. Various human monoclonal antibodies were introduced successfully in AIDP or refractory patients, however, further controlled studies are necessary [100,101].
Chronic Inflammatory Demyelinating Polyradiculoneuropathy
(CIDP)
CIDP is an uncommon progressive or relapsing paralyzing disease caused by inflammation of the peripheral nerves [12]. Neurologic symptoms are decreased sensation, diminished or absent reflexes, elevated cerebrospinal fluid level, and evidence of demyelination [13]. Cellular and humeral components of the immune system attack myelin on large peripheral nerve fibers in CIPD, leading to demyelination that manifests as weakness, numbers, paraesthesia, and sensory ataxia [102). As the disease progresses, axonal loss occurs secondary to demyelination and is associated with a poor prognosis [102,103] CIDP is an acquired disorder of the peripheral nervous system has probably an autoimmune pathogenesis. The nature of the responsible auto-antigens is unclear in most patients. The frequency of such antibodies is significantly greater in CIDP patients than in normal control subjects [104].
Recent clinical trials have confirmed the short-term efficacy of IVIG, prednisone and TPE. In the absence of better evidence about long-term efficacy, corticosteroids or IVIG are usually favored because of convenience. Benefit following introduction of azathioprine, cyclophosphamide, cyclosporine, other immunosuppressive agents, and interferon-β and –α and rituximab has been reported but randomized trials are needed to confirm these benefits [103,104].
Hughes et al. recommended in 2006 that the principle treatments are [105]:
1) intravenous immunoglobulin or corticosteroids should be considered in sensory and motor CIPD,
2) IVIG should be considered as the initial treatment in pure motor CIDP,
3) if IVIG and corticosteroids are ineffective TPE should be considered,
4) if the response is inadequate or the maintenance doses of the initial treatment are high, 5) if the response is inadequate or adding an immunosuppressant or immunomodulatory drugs could be considered, 6) combination treatments or adding multidisciplinary management should be considered.
In the guidelines on the use of TA in clinical practice-evidence- based approach from the AAC of the ASFA, the AIDP and CIPD have the category I with the RG 1A or 1 [12,13] (Table 2). The main etiology of AIDP is autoimmune antibody-mediated damage to the peripheral nerve myelin. Several controlled trials comparing TPE to supportive care alone indicate TPE treatment can accelerate motor recovery, decrease time on the ventilator, and speed attainment of other clinical milestones [12]. The Cochrane Neuromuscular Disease Group review of TPE in AIDP found that TPE is most effective when initiated within 7 days of disease onset. In recent years, IA has been increasingly recognized as an alternative to TPE for AIDP and CIPD [106].
Miller-Fisher syndrome (MFS) is characterized by the acute onset of ophthalmoplegia, ataxia, and areflexia. It is considered a variant form of Guillain-Barré syndrome. Because MFS is classified as a variant form of GBS and has a close association with the presence of the anti-GQ1b antibody, one would expect the efficacy of treatment with TPE or IVIG to have been proved. Anecdotal reports of the response of patients with MFS to TPE would be consistent with a pathogenic role for the anti-GQ1b antibody. However, there are some MFS patients without this antibody, and the ultimate proof that anti-GQ1b antibody mediates MFS has not been demonstrated [94]. MFS patients had deviated T-helper Type-1 (Th3) / T-helper Type- 2 (Th3) polarization and TPE can shift Th3-dominant status to Th- 1dominant status in patients with MFS. TPE may remove humoral factors including anti-GQ1b, and may induce a shift of the Th3/Th3 cytokine-producing cell balance in peripheral blood. Nowadays, there are case reports of GBS and MFS in Covid-19 patients and by the clinical suggestion of treating neurological complications with IVIG [107,108]. In the guidelines of the AAC of the ASFA, the Miller-Fisher syndrome has the category III and the RG 2C (Table 2) [12,13]. Further controlled studies would be useful.
Myasthenia Gravis (MG)
MG is a disease caused by autoantibodies, which are directed against acetylcholine receptors of the skeletal muscles. The acetylcholine receptor antibodies (Ach-R-ab) belong to a heterogenous group of polyclonal abs, which are directed against various sections of the postsynaptic receptor molecule. Due to blockage of the receptors, normal nerve transmission from motor nerves to striated muscle is interrupted. This disease primarily affects the muscles of the eyes, esophagus, and respiratory muscles, as well as the extremities.
Subgroups are patients with muscle-specific kinase (MuSK) and the low-density lipoprotein-related protein (LRP4) antibodies [109]. MuSK, a transmembrane tyrosine kinase, is expressed predominantly at the postsynaptic membrane of the neuromuscular junction (NMJ). MuSK binds LRP4 and transmits an agrin- mediated signal for the clustering of AChR [110]. MG with anti- MuSK antibodies corresponds to about of the MG patients. The LRP4 protein belongs to a family of proteins that has been recently identified as the receptor for the neural agrin that can activate MuSK [111].
The therapies are thymectomy and administration of cholinesterase blocking substances [112]. In cases with severe progression, immunosuppressives are also given to suppress autoantibody synthesis. TPE has been implemented with good results, especially in the case of severe, previously therapy-resistant progression [113]. The rapid elimination of autoantibodies achieved with TPE results in an improvement in clinical symptoms within hours to days. With the rapid improvement in the symptoms of their patients through TPE. Immunosuppressive drugs target autoantibody production but can take months to have an effect. IVIG and TPE have a more rapid effect than immunosuppressive therapy [114].
The rationale for TA is to remove circulating autoantibodies. In acute attacks, TPE is the first-line therapy (Table 2). The seropositve and seronegative patients respond to TPE. TPE is especially used in myasthenic crisis, perioperatively for thymectomy, or as an adjunct to other therapies to maintain optimal clinical status [12]. TPE works rapidly; clinical effect can be seen within 24 hours but may take a week. The benefits will likely subside in 2 – 4 weeks, if immunosuppressive therapies are not initiated to keep antibody levels from reforming. A combination of TPE and immunosuppressives seems to be successful. Dogra et al found TPE is high efficacious, cheaper short-term therapy for MG [115].
Rituximab, eculizumab, and belimumab, human monoclonal antibodies (HMA), are used in studies of patients with refractory MG and showed good results, but further studies are necessary, too [116,117].
Lambert-Eaten myasthenic syndrome (LEMS) is a rare, but reasonably well understood, antibody-mediated autoimmune disease that is caused by serum autoantibodies and results in muscle weakness and autonomic dysfunction [118]. Like MG, Lambert-Eaton syndrome is based on a disorder of the transmission of neuromuscular excitation. In these cases, no acetylcholine is released. LEMS is caused by an autoimmune attack against presynaptic voltage-gated calcium cannels and is characterized by late onset of fatigue, skeletal muscle weakness, weight loss, automatic dysfunction, and areflexia. It develops in the context of a malignant neoplasm, usually small cell lung carcinoma [119].
The rationale is similar to that in myasthenia gravis; that is, patient strength should be improved by the removal of the pathogenic antibody to the voltage-gated calcium channel. In most cases, patients are treated long-term with a combination of corticosteroids and immunosuppressive therapy has failed has TPE been attempted [120]. There are only case series, which have suggested some benefit by TPE. Further controlled studies must show the effectiveness.
Multiple Sclerosis (MS)
Multiple sclerosis is a replasing, remitting chronic demyelinating disease of the CNS and is the most common cause of neurologic disability in young adults [94]. Worldwide, there are more than one million afflicted with the disease. Alone in Germany, there are affected 120,000 to 140,000 patients with MS, and in the United States, there are more than 300,000 patients. MS is also diagnosed in children and adolescents. Estimates suggest that 8,000-10,000 children (up to 18 years old) in the USA have MS, and another 10,000-15,000 have experienced at least one symptom suggestive of MS.
The definition of MS as an autoimmune disease is based on the following characteristics [96]:– HLA association and genetic predisposition: T cell subset and cytokine correlation with disease activity, clinical responses to immunosuppression and immune activators, analogies with experimental autoimmune encephalomyelitis, cerebrospinal fluid oligoclonal IgG bands, CNS pathology using immunocytochemistry techniques, evidence of intrathecal synthesis of tumor necrosis factor beta in MS, and the level of TNF alpha in cerebro-spinal fluid may correlate with the severity and progression of disease and reflect histologic disease activity in MS, increased levels of gamma interferon correlate with the disease worsening.
MS is an autoimmune disease the pathogenesis is not clearly understood. TPE may be benefit MS patients by removing an antibody, such as antimyelin antibody, or by modulating immune response. There have been four immunopathologic patterns of demyelination in early MS lesions. The characteristics of demyelination for each pattern are [12]: T cell/macrophage-associated, antibody/ complement-associated, distal oligodendrogliopathy, and oligodendrocyte degeneration.
B-cells act as antigen-presenting cells to activate T-cells and produce proinflammatory (interleukin-6, interferon-γ, and tumor necrosis factor), and anti-inflammatory cytokines (interleukin-10) that regulate the immune process. These cells are also the source of mature plasma cells that secrete antibodies. Based on accumulation evidence, B cells participate in the pathogenesis of the disease through this multifunctional mechanism [121,122].
The rationale for treating MS patients with TPE derives from the presence of these circulating antimyelin antibodies, non-antibody demyelinating factors, aquaporin-4-specific serum autoantibodies, and neuroelectric blocking factors [123]. TPE removes antibodies and other humoral factors from the circulation safely and effectively. TPE has also been shown to increase the number and percentage of suppressor T cells and decrease the helper T cells in MS patients, thus effectively decreasing the ratio of elevated helper/inducer to suppressor/cytotoxic cell [124]. This point is important, because T cells play a pivotal role in the pathogenesis of MS [17]. TPE and IA, too, showed high efficacy and good tolerability [125]. Children should be treated with corticosteroids. If corticosteroids alone do not bring enough improvement, other treatments, including IVIG, Interferon ß 1a, and TPE, are available to treat-to-treat MS attacks. For drug removal in MS with natalizumab who develop progressive multifocal leukoencephalopathy (PML), TPE may also be used. PML is a severe opportunistic brain infection caused by virus, which is a known complication of natalizumab therapy [12].
In the guidelines of the AAC of the ASFA has acute attack of MS the category II and the RG 1A, 1B, the chronic MS and the chronic progressive MS the category III and the RG 1B respectively 2B (Table 2) [12,13]. The monoclonal antibody rituximab showed efficacy in the treatment of MS, although ocrelizumab, a humanized anti-CD20 antibody, showed beneficial effects on relapsing MS and partial effects on primary MS [126]. Other new anti-CD20 antibodies have been introduced in the treatment of MS: ofatumumab, and ublituximab, a new glycoengineered, chimeric anti-human CD20 [127]. However, further studies are necessary to see a benefit for patients with MS.
Pediatric Autoimmune Neuropsychiatric Disorders Associated with Streptococcal Infections (PANDAS), Sydenham´s chorea (SC)
PANDAS and SC are post-infectious neuropsychiatric disorders. Both have neuropsychiatric symptoms, which typically follow Group-A beta-hemolytic streptococcus (GABHS) infection. Streptococcal antigens induce antineural antibodies by an abnormal immune response if this pathogenesis is postulated [12]. GABHS infection has been associated with childhood-onset neuropsychiatric. The onsets of PANDAS are acute and dramatic which present with emotional/mood lability, attention deficit, deterioration of handwriting, separation anxiety, tactile/sensory defensiveness, enuresis, cognitive deficits, and motor hyperactivity [128].
SC is the main common acquired chorea of childhood. The major clinical manifestations are chorea, hypotonia, and emotional lability. The duration of SC is several months with a recurrence rate of about 20 percent [12]. The mean ages of onset for PANDAS and SC are 6.8 years and 8.4 years old, respectively. SC is diagnosed exclusively by clinical presentations and a history of rheumatic fever. Choreatic movements are rapid, and affect the face, trunk, and extremities. PANDAS are temporally associated with GABHS; it is not associated with rheumatic fever. Laboratory tests show elevated or increasing streptococcal antibody titers, but an elevated titer does not necessarily indicate a recent streptococcal infection. The presence of streptococcal infection in PANDAS is associated with at least two episodes of neuropsychiatric symptoms as well as negative throat culture or stable titers during times of remission.
The treatments for PANDAS include antibiotics and cognitive behavioral therapy. Severe form of SC is treated with diazepam, valproic acid, carbamazepine, or haloperidol [12]. If these fail, corticosteroids may be tried. While children with SC require long- term penicillin prophylaxis to reduce the risk of rheumatic carditis, the efficacy of penicillin prohylaxis in preventing symptom exacerbations in children with PANDAS remains doubtful. In severe symptomatic or refractory patients with PANDAS or SC, IVIG (1 g/kg/day for 2 days) or TPE has been shown to reduce symptom severity or shorten the course. TPE is indicated in severe extreme cases after the conservative therapy have been exhausted; or as first-line therapy in situations of life-threatening functional impairment [129]. The frequency is daily or every other day for five or six procedures over 7 to 14 days. There are no data on any benefit of repeated treatment. In the guidelines on the use of TPE from the AAC of the ASFA PANDAS or SC have the category I with RG 1B [12,13] (Table 2).
TA should be reserved for treatment of children and adolescents who are severely affected by PANDAS. In such patients, it appears to be safe, well-tolerated, and beneficial treatment option [130]. Bien et al reported in 2020, besides the first-line interventions of steroids, IVIG, and TA as second-line treatments cyclophosphamide or rituximab [131].
Chronic focal encephalitis (Rasmussen Disease)
The Rasmussen disease, is chronic focal encephalitis, and characterized by intractable focal seizures and slowly progressive neurological deterioration [12]. Onset is typically in childhood, mean age 6.8 ± 5.1 years, but a similar syndrome has been described in adults, too. The etiology of this disease is unknown, but antecedent infection with Eppstein-Barr virus, herpes simplex, enterovirus, or cytomegalovirus has been implicated. Cytomegalo- virus genome has been found in resected cortical tissue of three adult patients with Rasmussen´s encephalitis. Cerebrospinal fluid analysis in most cases is normal. Mild lymphocytic pleocytosis and elevated protein may be found. The important symptom of Rasmussen´s encephalitis is epilepsy uncontrollable with anticonvulsant drugs, progressive hemiparesis, and progressive unilateral cerebral atrophy. There is progressive loss of function in the affected cerebral hemisphere [12].
Anticonvulsants are necessary but are not always effective in controlling the disease nor do they stop its progression. Subtotal, functional complete hemispherectomy can markedly reduce seizure activity in a majority of patients but results in permanent contralateral hemiplegia corticosteroids and IVIG given for up to two years in a tapering schedule to diminish epilepsia and other symptoms [12].
Patients with Rasmussen encephalitis and antibodies against neural molecules, and autoantibodies can be produced in the CNS after cytotoxic T cell-mediated neuronal damage [13]. The Rasmussen encephalitis has the category III with RG 2C for TPE and IA in the AAC of the ASFA and the rationale for therapeutic apheresis is as follows:
Neuropsychological assessment may be helpful in evaluating patients with slowly progressive disease to determine whether TPE is effective in postponing surgical therapy. An initial course of TPE may be followed by 2 days of IVIG 1 g/kg/day. Monthly IA of 1.5 – 2 TPV per treatment has been reported effective in one patient [12]. Confirmation of anti-GluR3 antibodies may support the use of TA in patients with Rasmussen Ìs encephalitis. The frequency of TPE is every other day. After initial 5 – 6 TPE over 10 – 12 days, subsequent courses of TPE (with or without IVIG) may be performed at 2 – 3 month intervals as empirically needed. Immunosuppressive medications may increase the interval between courses. Until to date, there is no definitive consensus on treatment, with proposed strategies ranging from acute or chronic immunotherapy to hemispherectomy [132].
Acute Disseminated Encephalomyopathy (ADEM)
ADEM is an acute inflammatory monophasic demyelinating disease that effects the brain and spinal cord, which typically occurs after a febrile (often presumed to be viral) prodrome or vaccination [12]. Typical presentation for the multifocal neurological deficits is ataxia, weakness, dysarthria, and dysphagia accompanied by change in mental status. Most commonly, it is a monophasic illness that lasts from 2 to 4 weeks. Children and young adults are most affected. The differentiation of ADEM from the first attack of multiple sclerosis has prognostic and therapeutic implications. The features of ADEM, which can help to distinguish it from MS, are florid polysymptomatic presentation, lack of oligoclonal band in CSF, predominance of MRI lesions in the subcortical region with relative sparing of the periventricular area, and complete or partial resolution of MRI lesions during convalescence.
Corticosteroids are the first-line therapy, which hasten recovery and result in clinical improvement in up to 60 percent of patients. IVIG is for patients who do not respond to corticosteroids [12]. TPE is used and has a clearly defined role in other neurological conditions that are presumed to be immunologically mediated. TPE removes presumed offending antibodies as well as through immunomodulation. The category II for TPE with the RG 2C after the AAC of the ASFA is assigned on paucity of data [12,13] (Table 2). The frequency is every other day between 3 to 6 treatments. After Moussa et al. TPE appears to be of benefit for children with severe ADEM and warrants early consideration [133].
Other neurological diseases, such as cryoglobulinemic polyneuropathy, central nervous system systemic lupus, acquired neuromyotonia, polymyositis/dermatomyositis, polyneuropathy in paraproteinemia, neuropathy by hyperlipidemia and encephalopathy in metabolic/hematologic diseases such as thyrotoxicosis, hepatic coma, and M. Moschcowitz are diseases that involve more organ systems and are mentioned elsewhere. Extensive blood and plasma exchange for the treatment of the coagulopathy have been successfully implemented in children with meningococcemia [134]. Other TA methods like immunoadsorption or lymphocytapheresis have been applied in ataxic neuropathy and idiopathic hypertrophic cranial pachymeningitis, Fabry disease, acute transverse myelitis and subacute sclerotic panencephalitis with success [135,136].
Hematological Diseases
TPE is indicated in the management of various hematological diseases. Most medical management of immunohematological disorders requires the use of TA, serological immunomodulation, and classical pharmacological immunosuppression with steroids, cytotoxic agents, and antimetabolites, where overall therapy is individually tailored to the needs of the patient. Controlled trials are difficult if not impossible because of variables such as severity of disease, degree of organ system damage before intervention, age and the existence of co-morbid conditions. In some rare hematological diseases, it is impossible to recruit a large number of cases to perform a controlled clinical trial. Therefore, for most of these diseases only small series of cases are available for analysis.
TPE, semi-selective cascade filtration or IA aimed at the causative antibodies can be used in diseases caused by antibodies or ICs. Adjuvant drug therapies are different for various diseases and are typically individualized in type, dose and duration of use. The TA method chosen depends on the pathophysiological origin of a given disease. The physician who has chosen the TA method must be knowledgeable concerning the half-life time, the compartmental distribution of pathogenic plasma proteins, and the elimination of other toxic substances and complement components.
Rhesus Disease, Hemolytic Disease in Newborns (HDN)
Rh disease or incompatibility during pregnancy is an indication for TPE as a supportive therapy (12). Although it has been common practice for years to carry out anti-D gamma globulin prophylaxis in Rh-negative women after the birth of an Rh-positive child, increased anti-D antibodies still occur in up to 3% of subsequent pregnancies. This can lead to life-threatening morbus hemolyticus neonatorum for the fetus. Newborn babies rapidly develop anemia and hyperbilirubinemia with kernicterus. Exchange transfusion is the therapy of choice. Recently, TA has also become possible [137]. The diagnosis can be quickly made through the detection of anti-D antibodies in the mother and examination of the amniotic fluid for bilirubin and anti-D antibodies. Intrauterine exchange transfusions can be a lifesaving procedure but involve a high risk. The earlier Rh incompatibility manifests itself in pregnancy, the poorer the prognosis. If it occurs prior to the 26th week of pregnancy, more than 93% of fetuses die by the 31st week. If after the 26th week the Rh incompatibility manifests itself, and the mother receives TPE treatment, and the child receives intrauterine or postpartal exchange transfusion, 71% of these children can survive whereas without treatment most die [6].
Hemolytic disease in newborns presents as icterus neonatorum or hydrops fetalis. Both are caused by alloimmunization against RhD-positive red blood cells of a RhD-negative mother bearing an RhD positive fetus. Alloimmunization of the mother occurs after fetomaternal hemorrhage during the first pregnancy. The anti-RhD antibodies, which all belong to IgG subclasses, are able to transverse the placental barrier into the fetal circulation. The antibodies destroy fetal red blood cells by a no complement- dependent mechanism [6]. Hemolytic disease in newborns usually occurs during the second pregnancy with an RhD-positive fetus. Intravascular fetal transfusion with RhD-negative erythrocytes compatible with the mother’s serum is indicated in severe fetal hemolysis in a sensitized mother. After birth, the newborn may receive a phototherapy and/or a neonatal exchange transfusion, or TPE, depending on the severity of hemolytic disease in newborns [138].
The widespread use of fetal intravascular transfusion and the advent of IVIG therapy have now reduced the former significance of this disease. Towards the beginning of the second trimester in women who have, developed hydrops fetalis before the 22nd week of a previous pregnancy combined with IVIG, TA can be administered [139]. TPE with human albumin may bridge the gap between the onset of severe fetal anemia and the feasibility of fetal transfusion. To save the fetus for alloimmunization against other red cell antigens, which makes fetal intravascular transfusion impossible, maternal TA may be the only therapeutic option.Filbey et al. reported of 707 infants born to 583 alloimmunized women in Sweden [138]. Maternal TPE was performed in 2.4% of the cases with a response rate of 100%. TPE is recommended, therefore, only in severe HDN in the early stage of pregnancy before fetal transfusion is possible. TPE has been successfully performed thousands of times in recent years for Rhesus incompatibility. The physician must be aware that anti-D antibodies can also increase with TPE.
Bing et al. reported successfully treating 44 pregnant women with Rh incompatibility using a combination of anti-D immunoglobulin and TPE, and intrauterine transfusion. The effects gained from the therapy lasted for approximately 6 weeks for the patients. The study demonstrated that systematic management (including routine test for the presence or absence of D antigen in pregnant women, series test of anti-D antibody titer and ultrasonography, amniocentesis and cordocentesis) and timely treatment (including anti-D immunoglobulin, TPE, intrauterine transfusion, and delivery) could improve the perinatal outcomes of Rh-negative women [140]. TPE may delay the development of fetal anemia and reduce the risk of early and repeat intrauterine transfusion in cases with alloimmunization in pregnancy [141]. However, the application of TPE during pregnancy remains largely empiric and relies on individual case reports in the absence of high-quality studies and definitive evidence-based guidelines. Safety profile of TPE during pregnancy appears to be comparable to application of TPE in non-pregnant patients [142].
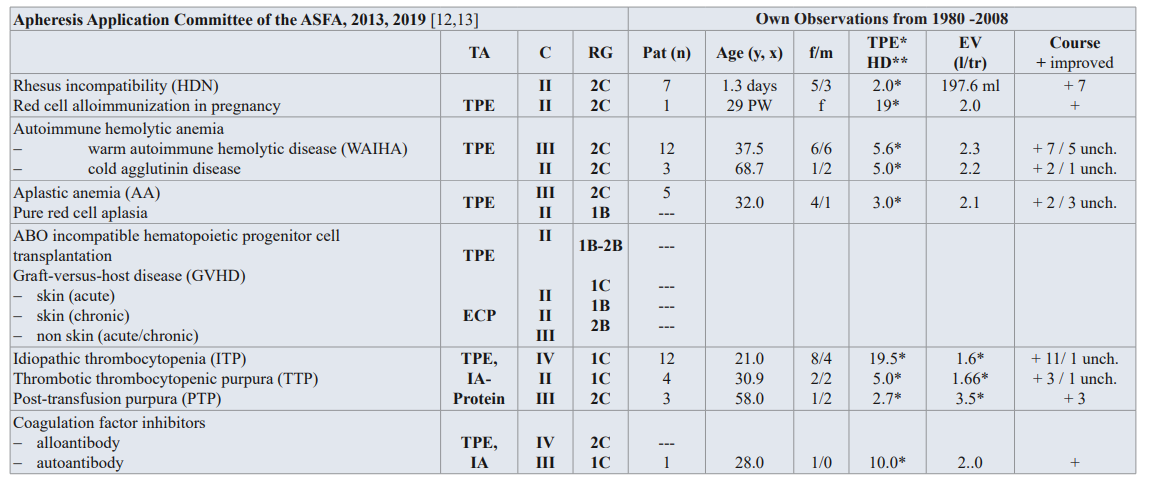
Table 3: Guidelines on the use of TA in clinical practice-based approach in hematological diseases with immunologic origin [12,13], and own observation from 1980-2008.
TA: TA modality; C: Category; RG: Recommendation grade; Pat: patient; f/m: female/male; EV: Exchange volume (l/tr: liter / treatment,x); Substitution: 3-5 % human albumin electrolyte solution, FFP; unch: unchanged. PW: pregnant week; ECP: extracorporeal photopheresis, IA: Immonadsorption on protein A
Category I: accepted for TA as first-line therapy; Category II: accepted for TA as second-line therapy; Category III: not accepted for TA, decision should be individualized; Category IV: not accepted for TA, approval is desirable if TA is undertaken [12,13].
The AAC of the ASFA has given HDN category II and RG 2C [12,13] (Table 3). The rationale for therapeutic apheresis is that TPE removes the maternal red cell alloantibodies that are responsible for HDN [12]. TPE can decrease the maternal antibody titer and, in turn, the amount transferred to the fetus, thereby protecting it from HDN. Survival in severe cases of HDN with the use of TPE and/or IVIG prior to ultrasound tomography (IUT) is about 70%. Category II for TPE is assigned for patients when there is a previous history of a severely affected pregnancy and the fetus is less than 20weeks gestational age [12]. Typically, IUT can be performed after the fetus reaches 20weeks of gestation.
TPE can safely be performed during pregnancy. During pregnancy, blood volume and especially the plasma volume increases. In the second or third trimester, it is preferable to place the patient on her left side to avoid compression of the inferior vena cava by the gravid uterus. Hypotension should be avoided as it may result in decreased perfusion to the fetus [137]. TPE should be considered early in pregnancy (from the 7th to 20th week) and continued until IUT can safely be administered (about 20th week of gestation). Close monitoring of the fetus for signs of hydrops will aid in guiding treatment. One approach is to use TPE for the first week (three procedures) followed by IVIG at 1g/kg weekly [12].
Hemolytic Anemia
The etiologies of hemolysis often are categorized as acquired or hereditary. Most acquired causes of hemolytic anemia are autoimmunity, microangiopathy, and infections. Immune-mediated hemolysis, caused by anti-erythrocyte antibodies, can be secondary to malignancies, autoimmune disorders, drugs, and transfusion reactions. When the red cell membrane is damaged in circulation, a microangiopathic hemolytic anemia is the consequence, leading to intravascular hemolysis and the appearance of shistocytes. Infectious agents such as malaria and babesiosis invade red blood cells [6].
The severity of hemolytic anemia is quite variable. Depending on the cause, it can be mild and compensated for by increased erythropoiesis. The treatment for mild forms and forms of such severity as to decrease red cell mass is directed at correction of the underlying cause. For example, proper antibiosis and supportive care for infections, surgical debridement and antibiotics for Clostridium welchii, and stopping the offending drugs in the case of G6PD deficiency. In severe hemolytic anemia, with hemoglobinemia, heme saturation of albumin and hemoglobinuria regardless of whether it is mediated by exogenous or endogenous noxae, timely implementation of TPE appears justified [137].
Autoimmune Hemolytic Anemia (AIHA)
AIHAs are characterized by reduced erythrocyte in vivo survival time and by the pressure of warm or cold agglutinizing antibodies against the autologous erythrocytes. Differentiation between the following antibodies is made based on their serological features [12,13]:
- Thermo-type: Warm agglutination autoantibodies. These autoantibodies consist mostly of IgG and its various Optimum antibody binding activitis is reached at body temperature (37°C).
- Cryo-type: Cold agglutination autoantibodies. These belong to the group of IgM antibodies and display their strongest reaction to antigen bearing cells at low temperatures (0–10°C). They become of clinical importance when a temperature of 30°C or more is reached.
- Bithermal autoantibodies: These belong to the IgG antibodies. Contrary to thermo-type, antibodies bind at low temperature (0–10°C) and hemolyse erythrocytes at body temperature (37°C) [143].
AIHA is diagnosed by direct microscopic evaluation of the peripheral blood film, hyperbilirubinemia, reticulocytosis, positive direct antiglobulin test (direct Coomb’s test), and elevated serum LDH [143,144]. Immune hemolytic anemia is a result of antibody fixation to a red cell antigen. This autoantibody triggers either intravascular red cell destruction mediated by the terminal lytic complement complex (C5b-C9) or extravascular destruction mediated by macrophage-phagocytic system [145]. Both mechanisms require opsonization by antibodies or C3b complement [146]. The antibodies mostly belong to the IgM (cryo- type abs) and IgG groups, or occasionally also to the IgA (thermo- type abs). The formation of the autoantibodies is still unknown.
When warm autoantibodies attach to red blood cell surface antigens, these IgG-coated red blood cells are partially ingested by the macrophages of the spleen, leaving microspherocytes, the characteristic cells of AIHA. Cold autoantibodies (IgM) temporarily bind to the red blood cell membrane, can activate complement, and deposit complement factor C3 on the cell surface. The macrophages of the liver (extravascular hemolysis) slowly clear these C3 coated red blood cells [143].
Although most cases of autoimmune hemolysis are idiopathic, potential causes should always be sought. Lymphoproliferative disorders (e.g., chronic lymphocyte leukemia, non-Hodgkin’s lymphoma) may produce warm or cold autoantibodies. A number of commonly prescribed drugs can induce production of both types of antibodies. Warm AIHA (WAIHA) also is associated with autoimmune disease (e.g., systemic lupus erythematosus), while cold AIHA may occur following infections, particularly infectious mononucleosis and Mycoplasma pneumoniae infection. Human immunodeficiency virus infection can induce both warm and cold AIHA [144]. Along with conventional therapy with corticosteroids and cytostatics or even splenoctomy, TA is increasingly being implemented with success [147].
The AAC of the ASFA has given autoimmune hemolytic anemia category III with RG 2C for the warm autoimmune hemolytic anemia and for the cold agglutinin disease category II with RG 2C [12,13] (Table 3). The observed symptoms include fatigue and jaundice. The laboratory findings are the signs of hemolysis such as anemia, hyperbilirubinemia, elevated serum LDH, reticulocytosis, as well as a positive direct antiglobulin (Coombs) test [12].
Prednisone is usually ineffective, as is splenectomy, because the liver is the dominant site of destruction of C3b-sensitized red cells [147]. TPE can remove effectively pathogenic immune complexes, activated complements, and autoantibodies [12]. The duration of the TPE treatment is until the hemolysis is controlled and the need for transfusions is limited.
Aplastic Anemia (AA)
Until now only some case reports of AA, which have been treated with TPE, are available. The pathogenesis of AA is regarded as complex and mostly unclear. In some cases, hemopoietic and erythropoietic inhibitors have been found in serum, leading to it being considered an autoimmune disease [6]. In these patients, it is possible to remove the circulating inhibitors by TPE. However, TPE is only indicated in the case of proven autoimmune pathogenesis. Successful therapy has also been conducted in recent years with cyclosporin A. The AAC of ASFA has given aplastic anemia and pure red cell aplasia (PRCA) category III with RG 2C [12,13,148] (Table 3). Aplastic anemia and pure red cell aplasia are rare hematopoietic stem cell disorders.
Allogenic hematopoietic progenitor cell (HPC) transplant is the treatment of choice for severe AA in newly diagnosed patients < 40years old. Young patients with mild disease or without a matched donor and older patients are treated with antithymocyte globulin, cyclosporine A and/or rituximab [149,150]. Immunosuppressive therapy is usually sufficient until remission is obtained in primary acquired PRCA. Corticosteroids (prednisone at 1mg/ kg per day) are used first. Alternative treatment is required if no response is achieved after 2–3months. Salvage agents include cyclophosphamide, azathioprine, cyclosporine, ATG, and high dose IVIG [151]. In diseases that may be immunologically mediated, TPE may be helpful by removing serum antibody and/ or inhibitory activity.
ABO Incompatible Hematopoietic Progenitor Cell Transplantation
The presence of natural antibodies in the recipient against the donor’s ABO blood group, which may cause hemolysis of red cells present in the transplanted product, is the requirement of the major incompatibility [13]. In peripheral hematopoietic progenitor cells that are collected by apheresis, there is a lower risk of hemolysis due to reduced red cell contamination (2–5%) as compared to HPCs derived from the bone marrow. To prevent either an acute hemolytic reaction the product needs to be red cell reduced or the patient’s antibody titer needs to be lowered. If the recipient has a high titer of antibodies, especially a group 0 patient receiving a group A transplant, a delayed erythroid engraftment or even pure red cell aplasia may result [12].
The AAC of the ASFA has given category II with RG 1B–2B for TPE in ABO incompatible hematopoietic allogenic progenitor stem cell transplantation (HSCT) and bone marrow transplants [12,13] (Table 3). TPE can reduce ABO antibodies, which are responsible for hemolysis and PRCA. In most of the ABO incompatibility,removal of the high titer antibody from the recipient’s circulation can prevent hemolysis if red cells are unable to deplete the product.
In minor incompatibility with passenger lymphocytes making antibodies 7–12 days after infusions, prophylactic red cell exchange with group O red cells can be performed to deplete recipient type red cells [12,13]. If unable to red cell deplete the HPC product, TPE should be performed before infusion of HPCs and the replacement fluid is a combination of albumin and plasma (50:50) compatible with both donor and recipient [12]. Before HPC transplantation, the goal should be to reduce the IgM or IgG antibody titers to ≤ 1:16 immediately. Generally, 2–4 TPEs are sufficient and if the antibody titer is high in the case of delayed red cell recovery or PRCA, TPE may be performed in the transplantation period [12]. ABO-antigen-specific IA was successfully introduced on ABO-incompatible hematopoietic cell transplantation [152] Immunosuppression, TPE, donor lymphocyte infusion, rituximab, and bortezomib, all with limited bebfit, and/ or daratumab demonstrate safety and high efficiency, suggesting its applicability as early treatment of post-allo-HSCT pure red cell anemia [153].
Graft-Versus-Host Disease (GVHD)
The GVHD has category II with RG 1B–2C for acute or chronic skin, and III with RG 2B for acute or chronic non-skin for extracorporeal photopheresis (ECP) after the AAC of the ASFA [12,13] (Table 3). GVHD following allogenic progenitor cell transplantation (HPCT) is typically characterized as either acute (aGVHD) or chronic (cGVHD) [12]. Acute GVHD usually occurs within 3 months after allogenic stem cell transplantation and results from activation of donor T cells by host antigen-presenting cells, leading to immune and cytokine-mediated tissue injury. The skin, gastrointestinal tract (GI), and liver are major targets of aGVHD. Chronic GVHD often evolves from aGVHD and is mediated by donor allo- or autoreactive T cells that activate inflammatory cytokines, B cells, autoantibody production, and cytolytic process. End-organ complications of cGVHD include progressive fibrosis and/or dysfunction of the skin, eyes, mouth, lungs, GI, joints, and vagina [12,13]. Acute GVHD of grades II to IV severity is first treated with a calcineurin inhibitor and systemic corticosteroids. Treatment options include local/topical measures for the skin, eyes, mouth, and gastrointestinal tract along with systemic therapies such as calcineurin inhibitors, ATG, mycophenolate mofetil, rapamycin, thalidomide, hydroxychloroquine, sirolimus, pentostatin, monoclonal antibodies against T cells, B cells or cytokines, and ECP [12].
The rationale of ECP involves the collection of peripheral blood leukocytes by apheresis, the extracorporeal exposure of the leukocytes to 8-methoxypsoralen (8-MOP) followed by irradiation with ultraviolet A (UVA) light, and the reinfusion of the photactivated cells [12]. The therapeutic effect of ECP for GVHD appears to involve induction of apoptosis in treated lymphocytes, modulation of monocytes-derived dendritic cell (DC) differentiation, increased production of anti-inflammatory cytokines by monocytes and T cells, decreased DC antigen- presenting function, restoration of normal T helper cell and DC subsets and induction of regulatory T cells that establish immune tolerance. For cGVHD, ECP improves skin or oral manifestations in 60–80% of steroid-dependent patients. Liver or GI complications respond in roughly 35–75% of cases, with the highest rates reported in children. Most responses with cGVHD are partial [12,13].
ECP is a well-established second-line-therapy for cGVHD. The role in the treatment of cGVHD is less clear but also points towards an effective second-line therapy option [154]. In future ECP could play a role in the prevention of GVHD. The treated volume is a mononuclear cell (MNC) product of approximately 270 mL consisting of mononuclear cells, plasma and saline [6]. The two-process method collects and treats MNC obtained from two times TPV processing. The replacement fluid is that all photo- activated leukocytes are reinfused with albumin and saline.
Thrombotic Thrombocytopenic Purpura (TTP)
TTP is a rare and life-threatening TMA syndrome [155], TTP is a systemic thrombotic illness, which is characterized by the only consistent abnormalities of microangiopathic hemolytic anemia and thrombocytopenia. Recently, TTP has been shown to be associated with a severe (< 5 percentage) deficiency of ADAMTS13 enzyme, which is a protease that cleaves multimers of von Willebrand factor. Idiopathic acquired TTP is associated with antibodies that bind ADAMTS13 and neutralize the protease activity [12]. Severe ADAMTDS13 deficiency appears to be an improvement proximal step in the pathophysiology of TTP. However, some patients with idiopathic TTP have no defect in ADAMTS13 function. Pregnancy, connective tissue disease (e.g., SLE), medications, infection, cancer, and transplantation are all associated with TTP and HUS syndrome [12].
TTP is a medical emergency, since the mortality of untreated patients exceeds 80% [156]. The mortality is thought to be caused by disseminated microvascular thrombosis, which may provoke ischemic injury and multiple organ failure. Ischemic organ failure can affect all organs, but the brain and heart are typically most affected. Acute kidney injury requiring dialysis and resulting in chronic kidney disease is rare. Central nervous system involvement is often manifested by transient focal neurologic deficits.
The mainstay of treatment is TPE with plasma replacement [157,158]. TPE works by removing antibodies against the von Willebrand factor cleaving protease, ADAMTS13. The plasma infused as part of the procedure also provides active ADAMTS13 protease to the patient, further restoring a more physiologic state of von Willebrand factor multimers. Other treatment modalities in non-responders to TPE include immune-suppression, not limited to high dose corticosteroids, and B cell depletion agents (e.g., rituximab). Patients with autoantibodies against ADAMTS13 do not always manifest TTP, and these antibodies alone are not sufficient to demonstrate the impending relapse of the disease. It is considered better to avoid platelet transfusion, which should be reserved for patients with clinically significant bleeding, as severe thrombocytopenia itself is not an indication for platelet transfusion. Conversely, platelet transfusion is probably not as dangerous as it was thought to be a decade ago [159].
The majority of acquired TTP in adults are related to an autoimmune disorder where anti-ADAMTS13 antibodies cause that deficiency and subsequent platelet adhesion and aggregation. Bacterial infections, autoimmune diseases, pregnancy, drugs, HIV infection, cancers, organ transplantation are the most frequent clinical conditions associated with TTP [13,160].
The first-line therapy in TTP includes corticosteroids, high dose intravenous immunoglobulin, cyclophosphamide, danazol, vinca alkaloids, and mycophenolate mofetil. The second-line therapy includes first TA, than HMAs, and/or thrombopoietin-receptor agonists [161]. The AAC of the ASFA has given TTP the category I and the recommendation grade 1A for TPE (Table 3) [12,13].
Idiopathic Thrombocytopenic Purpura (ITP)
ITP is an inherited or acquired disease that results in a reduction of circulating thrombocytes. This condition may be asymptomatic or manifests itself in hemorrhagic diathesis with petechial bleeding (161). The immune thrombocytopenias are a heterogenous group of bleeding disorders with similar hemostatic manifestations but different pathogenic etiologies. ITP caused by autoantibodies, which, in severely progressing cases, are accompanied by hemorrhagic diathesis. ITP is the most common autoimmune hematologic disorder. The etiology is still for the most unknown. The spleen plays an important role, since it not only produces a large part of the antibodies directed against thrombocytes, but also breaks down the damaged thrombocytes. As the antibodies can pass through the placenta barrier, the fetus can also be affected [162]. In more than 60 percent of the patients, part or full remission can be reached with corticoid steroid therapy. Splenectomy and cytostatics are further therapeutic measures. Acute and chronic cases have also successfully treated with high doses of intravenous immunoglobulin of 400 mg/kg BW/ day. In recent years, in addition to being treated with TPE, therapy resistant. The pathophysiological mechanism in ITP is the binding of auto- or alloantibodies to platelet antigens. Fixed antibodies may trigger complement activation [162]. The promised platelets are destroyed by phagocytosis in the macrophage-phagocytic system mediated by the Fc receptors FcRI-III and complement receptors CR1 and CR3. Platelet destruction occurs mainly in the spleen, and accessory spleen, but also in liver and bone marrow. The spleen is a major site of antiplatelet antibody production; therefore, splenectomy is therapeutically very effective. The main antigenic determinants are the platelet membrane glycoproteins GP-Iib/IIIa and Ib/IX [158].
A further mechanism leading to platelet destruction in drug-induced immune thrombocytopenic purpura is the formation of antibodies against neoantigens expressed after adherence of the drug to the RBC membrane [163-165]. Recently, acquired autoimmune deficiency of a plasma metalloprotease named ADAMTSJB was shown in many cases of ITP [166]. Alloimmunization is the cause of neonatal autoimmune thrombocytopenia, platelet transfusion refractoriness, and post-transplant purpura. The alloantigens are classified on the HPA system [167]. Neonatal immune thrombocytopenia is the platelet counterpart of hemolytic disease in newborns. QA HPA-Ia-negative is sensitized to HPA-1-positve platelets of the fetus. Alloimmunization (IgG ab > IgM ab) against platelet induced by fetomaternal hemorrhage occurs during a HPA-incompatible pregnancy or after a HPA-incompatible platelet transfusion. In heparin-induced thrombocytopenia, type II immune complexes consisting of antibodies to heparin and platelet factor 4 activate platelets after binding to platelet Fc receptors. Excess platelet factor 4 binds to endothelial glycosaminoglycan, resulting in endothelial damage and thrombi [156]. Heparin- induced thrombocytopenia type I refers to non-immunogenic thrombocytopenia due to heparin-induced aggregation of platelets.
Acute abrupt onset ITP is seen in childhood, and often follows a viral illness or immunization. The majority of children requires no treatment and in 80–85 percent of cases the disorders resolve within 6 months. Some 15 – 20 percent of children develop a chronic form of ITP, which in some cases, resembles the more typical adult. Chronic ITP in childhood has an estimated incidence of 4.6 per 100.000 children per year and prevalence of 4.6 per 100.000 children at any one time [156]. This form of ITP affects mainly women of childhood age (female: male: 3:1). Childhood ITP has an incidence of between 4.0 and 5.3 per 100.000 [159].
The diagnosis of ITP based principally on blood count, clinical symptoms, autoimmune profile and other investigation, and on the exclusion of other causes of thrombocytopenia using the history, physical examination. Further investigations are not indicated, blood count and film are typical of the diagnosis of ITP and do not include unusual features that are uncommon in ITP [156]. Platelet associated IgG (PAIg) is elevated in both immune and non-immune thrombocytopenia and therefore has no role in the diagnosis of uncomplicated ITP. In patient’s refractory to therapy although some patients have shown improvement in platelet counts following eradication therapy, it is worth determining the presence of H. pylori.
The successful use high doses of IgG and anti-D therapy have reduced TA, second-line treatment in these cases [156]. The first-line therapy is splenectomy and high doses corticosteroids, high dose IVIG, intravenous anti-D, cyclosporin A and dapsone. Patients who failed the first-line therapies must be treated with interferon (IFNα), rituximab, campath-1H, mycophenolate mofetil and TA [162]. TA can include remissions in approximately 80 percent of patients with ITP. TA becomes a legitimize option for maintenance therapy in chronic ITP patients, if the application of IgG is not possible due to allergic reactions, Rh-negative status, or splenectomy.
The most important part of TA is to remove anti-platelet antibodies to prevent bleeding by keeping the platelet count above a critical level. The goal of therapy is to obtain sustained remission with a minimum platelet count of over 50.000 platelet/ μl. As some severely progressing cases of ITP do not respond to steroids and/or high doses of immunoglobulin, immunosuppressive drugs, to four sections of TPE per month and biologics, TPE is indicated [163].
As there are only a few controlled studies yet available, it is not possible conclude which form of therapy should be given preference. Thus, in ITP, initial treatment should consist of corticoid steroids, high doses of IgG, and immunosuppressive drugs as mentioned above. Should no significant improvement be observed within one or two weeks (thrombocytes > 80.000 μl), then TA treatment should be commenced immediately. The authors recommend TPE with 1 to 1.5 plasma volume a day for 4 days, and to four sections of TPE per month have a positive effect in chronic cases. TPE is recommended prior to surgery in acute respectively chronic uncontrollable bleeding [156]. IA with Protein-A was also induced successfully in the treatment of ITP [164].
In the AAC of the ASFA, ITP has the category III and the RG 2C for TPE and IA in refractory cases (Table 3) [12,13]. First-line therapies are oral corticosteroids, IVIG (1-2 mg of prednisolone/ kg/day, IVIG at 1 g/kg for 1-2 days), and is anti-Rh (D) (50-74 μg/ kg) [12]. If thrombocytopenia persists or recurs, splenectomy is recommended in adults but is differed to prevent overwhelming post splenectomy infection or allow for spontaneous remission. TPE and IA may have been considered in patients with refractory ITP, with life-threatening bleeding or in whom splenectomy is contraindicated. IgG antibodies and IgG-containing circulating immune complexes can be selectively removed by IA with protein-A. The use of this column is contraindicated when the patient is on ACE inhibitors, has a history of hypercoagulability, or thromboembolic events [165]. There are no clear guidelines concerning treatment schedule and duration of treatment. The procedure is generally discontinued when either the patients show improvement in platelet count > 50 x 10’/L or no improvement after about six treatments. The columns with protein-A are no longer available in the USA but may be available in other countries [12].
Great progress has been made in recent years in developing new treatment options for thrombocytopenic patients, especially in ITP. In addition to TA, human monoclonal antibodies and thrombopoietin agents, combination of different diagnosis and therapeutic approaches are the main strategy for difficult cases [156]. Especially rituximab is a novel second-line agent for the treatment of ITP with encouraged results of some studies. Thrombopoietin (TPO) is the major physiology regulator of platelet production. TPO is produced by the liver at a constant rate and cleared from the circulation by TPO receptors on circulating platelets thereby providing an efficient feedback system regulating platelet production by bone marrow megakaryocytes [166]. The further new second-line drugs for ITP are the thrombopoietin receptor agonists (TPO-RAs). For platelet response, elrombopag, and romiplostim were the best. Romiplostim and electrombag have high efficacy and safety as second-line treatments in the short term for adult patients with persistent ITP. Eltrombpopag and romiplostim stimulate the platelet and megakaryocyte production. Both substances are approved in the USA and the EU for treatment of different forms of thrombocytopenia [167].
Post-Transfusion Purpura (PTP)
PTP occurs when donor B lymphocytes and dendritic cells migrated as passenger’s cells to the recipients system, where they undergo clonal expansion after “homing in” on, and producing alloantibodies to the incompatible HPA allele [168]. PTP is rare bleeding disorder caused by alloantibody specific to platelet antigens. The antibody against the human platelet alloantigen HPA-Ia is responsible for the most of the cases. The majority of affected patients are multiparous women who presumably have been previously sensitized during pregnancy [169]. Blood transfusions rarely have been implicated as the primary cause for alloimmunization in PTP. Thrombocytopenia is usually severe and resolves spontaneously within several weeks. However, patients may develop severe if not fatal bleeding during the course of the disease. The diagnosis is confirmed by demonstrating that the patient´s serum contains antibodies to platelet-specific antigens.
The treatment is high IVIG (0.4 g/kg BW/day for 2-5 days or 1g/ kg BW/day for 2 days) [12]. It is possible acts by Fc receptor blockade of RES. The removal of HPA 1a alloantibodies by TPE results in a decrease of antibody titer, removal of any unattached HPA-1a antigen, and an increase in platelet count and cessation of bleeding. TPE should be considered as the urgent treatment of hemorrhage and severe thrombocytopenia if IVIG therapy is not effective [12]. In the AAC of the ASFA the PTP has the category III with the RG 2C for TPE based on limited data available in the literature (Table 3) [12,13]. TPE can discontinue when platelet count starts increasing (> 20 x 10â¹/L) and non-cutaneous bleeding stops.
Hemophilia A
This is a defect of the endogenous coagulation system, either inherited or acquired. It includes diseases that result from reduction, lack, or malformation of the factors VIII, IX, XI, XII, or prekallikrein. Hemophilia A is the longest-known hemorrhagic diathesis. Because of substitution therapy, 5–20% of hemophiliacs develop antibodies against factor VIII administered during the course of treatment. Factor VIII antibodies belong to the IgG immunoglobulin group [170,171]. Antibodies can, however, also occur spontaneously in older patients or after pregnancy. These antibodies are directed against the patient’s own factor VIII and can lead to an acquired factor VIII deficiency. Hemophiliacs may become sensitized to concentrates of their deficient coagulation factors. This occurs in about 15% of hemophilic patients. Low and high responders can be distinguished. The activity of the inhibitor can be measured in Bethesda units (BM) or Malmö inhibitor units (MiU). The F VIII inhibitors are IgG subclass 4 antibodies. F VIII inhibitors are the most common pathogenic antibodies directed against the blood coagulation factors. They develop in approximately 30% of patients with severe and moderately severe hemophilia A in response to infusions of F VIII. Patients develop inhibitors usually within the first year of treatment. The mechanisms underlying the state of apparent immune tolerance in the remaining non-inhibitor patients are unknown. The greatest risk of inhibitor development is associated with nonsense mutations, large deletions and intrachromosomal recombinations (inversion) in the F VIII gene that are predicted to cause a complete lack of endogenous F VIII. The risk of inhibitor development in patients with mild hemophilia A increases with the amount of exposure F VIII [172].
Many patients with antibody formation display a rapid increase in antibodies after administration of factor VIII. Attempts to suppress the formation of antibodies in these patients through immunosuppressive therapy have, for the most part, been unsuccessful. TA is used to reduce these antibodies prior to infusing factor VIII. TA in combination with factor VIII has been successful in interrupting severe bleeding in hemophilics who are unresponsive to Factor VIII and as hematologic preparation to normalize these inhibitors prior to major surgery [173].
TA is indicated in severely bleeding patients classified as immunological high responders [173]. TA can be considered when plasma concentration of the inhibitors exceeds either 10 BM or 3 MiU. TA should be implemented prior to high-dose administration of human VIII concentrates. The use of IA with anti-immunoglobulin columns may be safer and more effective. A further indication for TA is in cases where inhibitors occur after factor substitution to induce immune tolerance according to the Malmö or similar protocols. Serial TPE and simultaneous administration of factor VIII/IX concentrates, high dose IgG (0.4g/kg per day), and cyclophosphamide is recommended. This protocol has a success rate of 80%. Chronic immunosuppression may be necessary in some cases [173]. IA is being increasingly applied in the treatment of F VIII inhibitors. Several types of IA methods have been used. However, IA may be clinically effective and cost-effective and should be considered early in the treatment of patients [174] (Table 3).
Acquired Factor VIII (F VIII) antibodies in non-hemophiliac patients
Antibodies against factor VIII can occur in many diseases such as immunological diseases, after pregnancy, as a reaction to medication (e.g., phenylbutazone), skin complaints, tumors, and diabetes mellitus. In the case of most patients with acquired factor VIII antibodies, it is not possible to determine the cause. If the underlying disease is known and treated, a drop in antibody titer can be expected [6]. F VIII autoantibodies in non-hemophiliacs produce a condition sometimes called acquired hemophilia A. It is the most common autoimmune bleeding disorder involving the coagulation system. For unknown reasons, acquired hemophilia A patients are more likely to have a more severe bleeding diathesis than hemophilia A inhibitor patients. Approximately 50% of acquired hemophilia A patients have underlying conditions, including autoimmune disorders, malignancy, and pregnancy [175]. The remaining idiopathic cases most commonly occur in elderly patients of either sex.
Treatment of bleeding episodes for patients with acquired hemophilia A or congenital hemophilia A with inhibitors depends on the inhibitor titer. Low-titer inhibitors can be overwhelmed with F VIII bypassing agents (prothrombin complex concentrates, activated prothrombin complex concentrates), or recombinant F VIIa or porcine F VIII concentrates can be used to treat patients with high-titer inhibitors. Recombinant F VIIa is effective in controlling most bleeding episodes. There have been no reports of inhibitory antibodies developing to the product [176].
Acute bleeding complications are an indication not only for the application of highly dosed concentrated factor VIII, but also for the removal of circulating antibodies through TPE. Substitution with fresh frozen plasma also includes the administration of F VIII. The advantage of TPE and IA is in its rapid removal of antibodies and absence of excessive antibody formation. A disadvantage is an increased risk of bleeding with TPE treatment, if anticoagulation becomes necessary.
In the ASFA guidelines on the use of TA, the coagulation factor inhibitors in hemophilia A and acquired factor antibodies in non- hemophilia patients has category III with RG 2B for IA and IV with RG 2C for TPE [12,13] (Table 3). Factor deficiency can be either congenital or acquired; the majority of acquired deficiencies result from autoantibodies. In addition, congenital factor deficient patients can develop inhibitors, alloantibodies, to the factors. The treatment options for inhibitor suppression include high dose corticosteroids, cyclophosphamide, cyclosporin, rituximab, and high dose IVIG [6]. For coagulation factor inhibitors, the extracorporeal removal by IA is more effective than TPE [176].
Since the beginning of gene therapy, hemophilia has been considered an attractive disease target that serve as a trailblazer for the field at large. The most promising vectors for hemophilia gene therapy are adeno-associated viral vectors (AAVs) and lentiviral vectors. More recently, gene-editing approaches based on designer nucleases or CRISPR/Cas, have also been considered to minimize risks associated with random vector integration and insertional mutagenesis though off-target issues would have to be carefully and comprehensively assessed [177]. In patients with high titer of antibodies is the use of some TPEs or IAs before the introduce of the gene therapy could be discussed.
Dermatological diseases
Dermatologic immune mediated diseases represent a heterogenous group of disorders associated with circulating autoantibodies against distinct adhesion molecules of the skin and/or mucosa [178]. According to the level of split formation, the disorders can be divided in the intraepidermal blistering pemphigus, such as pemphigus vulgaris (PV), pemphigus foliaceus, and paraneoplastic pemphigus, and the subepidermal blistering pemphigoid diseases, such as bullous pemphigoid (BP), pemphigoid gestations, and dermatitis herpetiformis [12]. The new developed sensitive and specific assays for circulating autoantibodies in these dermatological diseases now enable a serological diagnosis in about 90% of cases.
The incidences of autoimmune blistering skin diseases in Germany has doubled in the last 10 years, to about 25 new cases per million humans per year, because of improved diagnostic techniques as well as the aging of the population [179]. There are an estimated 2000 new cases of autoimmune blistering skin diseases per year. The incidence of pemphigus in Europe is one to two cases per million humans per year, and 80% have PV. BP is the most common type of subepidermal autoimmune blistering skin disease in Europe, with an incidence of about 13 cases per million humans per year [180,181]. The next common types are mucous membrane pemphigoid and pemphigoid gestationis [182]. The standard of diagnostic testing for autoimmune blistering skin diseases is direct immunofluorescence (IF) microscopy to demonstrate the presence of tissue-bound autoantibodies and/or of C3 in patients’ skin or mucous membranes.
Pemphigus Vulgaris (PV)
PV is a severe, chronic disease of the skin and mucous membranes, has poor prognosis and acantholytic blisters and erosion, and is characterized by the presence of antibodies against epidermal intercellular substances [183]. Both genders are equally affected with the mean age of onset in the sixth and seventh decade of life, and the patients present with skin lesions that occur typically as flaccid blisters [13]. The blisters can be located on the entire body surface as well as on the mucous membranes of the mouth. PV has a high morbidity and mortality before the introduction of corticosteroids. They reduced the mortality rate from 70% to 100% to a mean of 30%. The conservative therapy include high doses of corticosteroids, dapsone, gold, and systemic antibodies alone or in combination with other immunosuppressant agents in usually dosages, such as azathioprine, methotrexate, and cyclophosphamide. Newer therapeutic modalities are mycophenolate mofetil, chlorambucil, dexamethasone-cyclophosphamide, IVIG therapy, TPE, ECP and rituximab [13,184]. The rationale for using TPE in the treatment of PV based on the presence of circulating pathogenic autoantibodies (Table 4). The frequency of TPE and IA is 3 to 6 treatments in two weeks, and then after the titer of the antibodies in the blood. For ECP, the frequency is 3 to 4 treatments in one week, and then after the antibody titers. The treated volume is for all diseases 1 to 1.5 TPV. The substitution solution for TPE is usually a 5 % human albumin-electrolyte solution or a part of FFP for all diseases. The goal of TPE is to reduce the level of autoantibodies with subsequent improvement in clinical symptoms. The decline autoantibody titers, antikeratinocyte cell surface antibodies, and anti–desmoglein-3 correlated with clinical response in a number of patients [178]. The antiepidermal antibodies, which usually belong to the IgG category, can be easily eliminated with TPE [185,186].
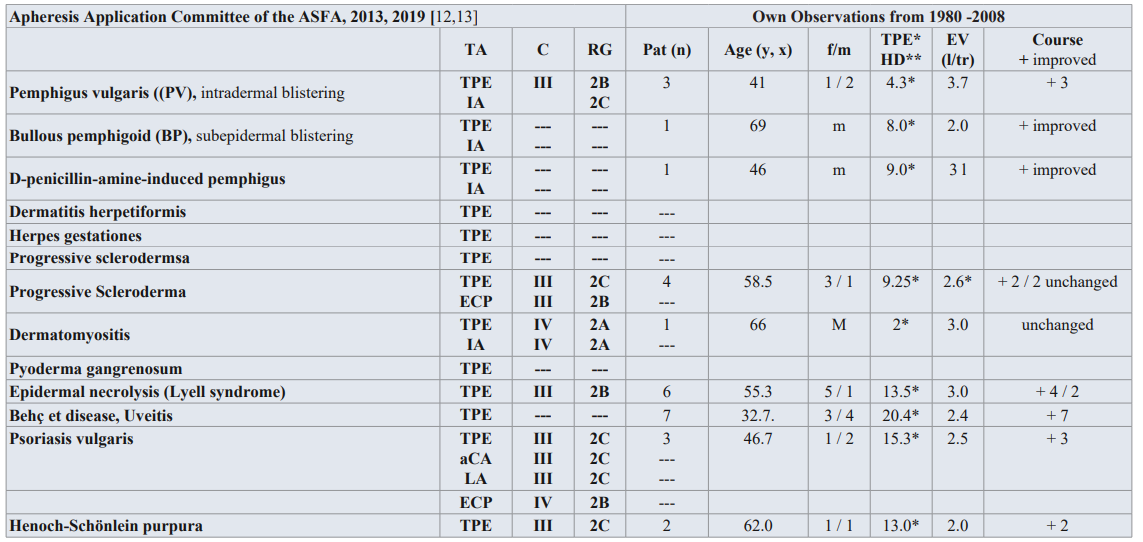
Table 4: Guidelines on the use of TA in clinical practice-based approach in dermatological diseases with immunologic origin [12,13], and own observation from 1980-2008.
TA: TA modality; C: Category; RG: Recommendation grade; Pat: patient; f/m: female/male; EV: Exchange volume (l/tr: liter / treatment,x); Substitution: 3-5 % human albumin electrolyte solution, FFP; unch: unchanged. aCA: adsorptive Cytapheresis, LA: Lymphocytapheresis, ECP: extracorporeal photopheresis; IA: Immunadsorption on protein A
Category I: accepted for TA as first-line therapy; Category II: accepted for TA as second-line therapy; Category III: not accepted for TA, decision should be individualized; Category IV: not accepted for TA, approval is desirable if TA is undertaken [12,13].
Bullous Pemphigoid (BP)
BP is another form of subepidermal blistering pemphigus; BP is rare. BP frequently involves a premonitory stage with pruritic urticarial erythema and eczematous lesions followed by the classical bullous stage with tense blisters, erosions, and crusts [186]. BP is a chronic dermatosis often associated with acute exacerbations, with the formation of bullae blisters usually on the inflamed skin, subepidermal blister formation, and antibodies against the epidermal basal membrane. Thus, BP can also occur in combination with other autoimmune disorders. The course of this pemphigus disorder is not as dramatic as other forms of the disease, with good response to high-potency corticosteroids, which are usually combined with dapsone, doxycycline, methotrexate, or azathioprine in usually dosages [179]. BP has an annual incidence of about 13 - 42 new cases per 1 million in central Europe and the United Kingdom [187,188]. Only a few cases have been treated with TPE up to now [189]. After 3 to 5 treatments in one week, we could see if TPE or IA can decrease the antibody titer. In very low antibody titers, the frequency of the treatments can decrease, too. Because the pathogenic relevance of autoantibodies therapeutic options was clearly demonstrated in the majority of autoimmune bullous diseases, removal of autoantibodies, therefore TPE is indicated IA and rituximab have been established additional therapeutic options [190].
D-Penicillamine induced Pemphigus
D-Penicillamine induced pemphigus, steroid-resistant pemphigus, is a foliaceus type disease with high lethality and mortality rate, which can occur as a side effect in long-term penicillamine therapy, which is a particular indication for TPE [191]. Only case reports of D-penicillamine-induced pemphigus treated successfully with TA were reported in combination with immunosuppression [192]. IA is the most specific therapeutic option, in which only the pathogenic IgG is depleted in the patient’s plasma. Three to 5 treatments of TPE or IA and immunosuppressive drugs in one or two weeks are necessary to an improvement of the disease. A combination of IA and rituximab showed rapid and long-lasting response of concomitant immunosuppressive medication [16,183]. Rituximab, in usually dosages, is almost given as an adjuvant drug, i.e., in addition to another type of immunosuppressive treatment. Complications of rituximab in patients with autoimmune blistering skin diseases include infections, deep venous thrombosis of the lower limbs, pulmonary embolism, longterm hypogammaglobulinemia, and neutropenia with an overall mortality of 4% [181]. The treatment with corticosteroids and cyclosporin is documented as first-line therapy. In cases that do not respond to this treatment, alternative therapeutic procedures (e.g., systemic corticosteroids and mycophenolate mofetil; mycophenolate mofetil and cyclosporin; tacrolimus; infliximab in usually dosages, or TPE [181].
TPE is in combination with immunosuppression probably successful due to the pathogenesis of severe cases of dermatitis herpetiformis and herpes gestationis [192,193]. Herpes gestationis or pemphigoid gestationis is an autoimmune subepidermal blistering disease that occurs in women in the second or third trimesters of pregnancy or even puerperium. It is a rare skin disease, the incidence of which has been estimated of approximately one case in every 40,000–60,000 pregnancies [193].
Scleroderma or systemic sclerosis is a rare, generalized autoimmune disease. Scleroderma is characterized by vascular abnormalities, fibrosis, inflammatory changes, and late-stage atrophy/obliterative vasculopathy. Localized scleroderma forms show a longitudinal or circumscribed skin involvement [194]. The effectiveness of TPE in progressive scleroderma and dermatomyositis is still disputed.
Pyoderma gangrenosum (PG) is a rare, polyetiological syndrome based on a pathological immune reaction. In over 40% of cases, this disease occurs together with colitis ulcerosa. In the vessel, walls of vasculitic lesions, granular IgG, C3, complement, and IgM deposits have been observed [195]. PG is a non-infectious neutrophilic dermatosis that usually starts with sterile pustules that rapidly progress to painful ulcers of variable depth and size with undermined violaceous borders. In 17 % -74 %of cases, PG is associated with an underlying disease, most commonly inflammatory bowel disease, rheumatological or hematological disease, or malignancy. PG is characterized by painful, enlarging necrotic ulcers with bluish undermined borders surrounded by an advancing zone of erythema; its clinical variants include ulcerative or classic, pustular, bullous or typical, vegetative, peristomal, and drug-induced. It can be idiopathic or associated with cancer, infections, medications, and systemic diseases [196]. The treatment with corticosteroids and cyclosporin is documented as first-line therapy. In cases that do respond to this treatment, alternative therapeutic procedures (e.g., systemic corticosteroids, and mycophenolate mofetil; mycophenolate mofetil and cyclosporine; tacrolimus, infliximab in usually dosage, or TPE (8-10 treatments) are recommended [197].
Drug-induced Epidermal Toxic Necrolysis (TEN),
TEN, also known as Lyell’s syndrome, is a life-threatening drug reaction characterized by extensive destruction of the epidermis and mucosal epithelia. The eyes are typically involved in TEN. The disease has a high mortality rate. TEN and the Stevens - Johnson syndrome (SJS) are closely related, although their severity and outcome are different. The SJS and TEN are rare but present severe skin manifestation. They are estimated to occur in one to three people per million per year in Europe and the United States (198). They are characterized by a low incidence but high mortality, and drugs are most commonly implicated in 80% of TEN cases. TEN is the most severe form of drug induced skin reaction and is defined as epidermal detachment of 30% of total body surface area [199]. In Lyell’s syndrome, the acute phase can be very successfully treated by TPE. The allergic or toxin-induced skin necrolysis is usually triggered by a drug acting like a hapten [200]. Lyell’s syndrome is fortunately very rare but has a high mortality rate, approximately 50%, and thus, early administration of TPE is justified, 6 to (8 to 10 treatments) every day or every other day. TPE is a safe intervention in severely ill TEN patients and may reduce the mortality in this severe disease [201].
Behçet disease a multisystem inflammatory disorder, presents with the involvement of muco-cutaneous, ocular, vascular, central nervous and gastrointestinal systems. It is an idiopathic, chronic, and recurrent disease characterized by exacerbation alternating with plasma of quiescence, episodic pan uveitis, and aggressive no granulomatous occlusive vasculitis of the arteries and veins of any size with explosive ocular inflammatory attacks that primarily affect the retinal and anterior segment vasculature of the eye [202]. Central nervous system involvement, most often due to necrotizing vasculitis, may be the most protein manifestation of the disease, leading to death. The frequency of ocular manifestations is 70%- 85% in these patients.
Although TPE has been has been successful in individual cases [203]. The frequency is 3 to 6 treatments daily or every other day, a chronic treatment of TPE with one treatment every two or four weeks for two or three months is possible, too. In recent years, there have been reports on the successful treatment with implementation of cyclosporin A, tacrolimus, or infliximab, etc.
Psoriasis vulgaris is a common autoimmune chronic inflammatory skin disease that affects approximately 2% of the mechanism is the secretion of type 1 (Th 1) cytokines by T cells and their activation [204]. TPE may be beneficial in patients with psoriatic arthropathy and not responding to conventional therapy [205]. However, blocking TNF-α by infliximab or etanercept has shown particular promise, especially in the management of psoriasis.
Henoch-Schönlein purpura (HSP) is a systemic vasculitis that affects vessels of small size. The vascular purpura is usually confined to the lower limbs and is associated, at varying degrees, with joint, gastrointestinal, and renal involvement. It is a systemic disease where antigen-antibody (IgA) complexes activate the alternate complement pathway, resulting in inflammation and small-vessel vasculitis [206]. HSP is defined as the presence of two or more of the following criteria: age of disease onset (20 years or younger), palpable purpura, acute abdominal pain, and granulocytic infiltration in the walls of arterioles or venuoles. All patients develop palpable purpura. In the skin, these deposits lead to subepidermal hemorrhage and small-vessel necrotizing vasculitis producing the purpura [13]. IgG autoantibodies directed at mesangial antigens may play a role in pathogenesis. In other organs, necrotizing vasculitis leads to organ dysfunction or hemorrhage. The recommendation for TPE are severe cases (Table 4). Eight to 10 treatments daily or every other day until the antibodies disappeared.
Autoimmune Diseases
The terms “systemic autoimmune disease” and “collagen vascular disease” describe a number of illnesses, the common characteristic of which is immune-mediated destruction of intracellular structures in connective tissue, resulting in fibrinoid tissue damage [207]. Based on an immune pathogenesis, the various organs form antigen components, which provoke formation of autoantibodies on the one hand, and circulating immune complexes causing inflammation in organ tissues on the other.
Antinuclear antibodies are to be found against most nuclear structures. The antibodies are typically directed against both cytoplasmic-associated and cell membrane-associated proteins, and antibodies against cytoplasmic structures and cell membrane components. The different groups of antibodies observed in active and subclinical disease includes those against many extracellular antigens, such as collagen, myelin sheaths, immunoglobulins, basement membrane, intercellular bridges, hormones, and complement components [6].
Systemic Lupus Erythematosus (SLE)
SLE is an autoimmune disease characterized by hypergammaglobulinemia, the presence of various autoantibodies, and immunoregulatory alteration. Among the autoantibodies, anti- double-stranded (ds) DNA is highly specific for the disease and is thought to play an important role in its pathogenesis. Anti-ds DNA autoantibodies constitute a heterogeneous family with respect to avidity, cationic charge, immunoglobulin class, and complement- fixing ability [6].
SLE usually involves high-titer antinuclear antibodies of the IgG group. This antinuclear antibody group includes not only the anti-ds DNA antibodies but also autoantibodies against single- stranded DNA (ssDNA), histones (h3–H4), and non-histone proteins (e.g., Sm, nRNP, SS-A/Ro, SS-B/La) [16]. Thus, in addition to antinuclear antibodies, SLE patients possess, although less frequently, autoantibodies against cytoplasmic antigens (SS- A/Ro, SS-B/La, ribosomes, and Golgi apparatus), phospholipids (e.g., cardiolipin), cytoskeletal proteins (e.g., cytokeratin, desmin, vimentin, and neurofilaments), basement membrane, and various cell superior determinants of leukocytes, erythrocytes, and thrombocytes.
Most antibodies belong to the so-called easy antigens, i.e., they are long-chained structures with repetitive epitopes, such as DNA, RNA-cell surface antibody, and basement membrane [6]. Many of these antibodies are polyreactive, i.e., show overlapping binding specificity for several antigens. The cause for the polyreactivity of anti-DNA antibodies is thought to be related to the fact that the various antigens have in common certain phosphate remains in a similar conformation or that the antigen-binding site of autoantibodies has various independent binding sites [6].
It is still unclear whether formation of antinuclear antibodies is due to altered (and thus immunogenic) DNA, polyclonal activation of B cells, cross-reaction with bacterial antigens, or a genetically induced disorder of immune regulation. The importance of genetic factors is not only underscored by the abovementioned relationship between human leukocytes and antibodies, but also by recent immunologic analyses of anti-DNA autoantibodies in mice and humans. Natural CD4+ CD25+ regulatory T cells (Tregs) have a potent immunosuppressive function and contribute to immunologic self-tolerance by suppressing potentially autoreactive T cells. Depletion of these cells leads to destruction of severe autoimmune diseases in animal models; more recently, there have been studies reporting impairment of Treg numbers and/or function in various human autoimmune diseases [208]. The tissue damage is caused by deposition of circulating immune complexes in various organs. Primarily involved are the smaller and medium-sized arteries of the skin, joints, lungs, liver, brain, kidneys, glomeruli, peritubular renal capillaries, and epidermal basement membrane [2].
SLE is a chronic inflammatory disorder. With its extremely variable range of symptoms, SLE can cause broadly varying clinical conditions, ranging from an acute attack with high temperature, anemia, leukopenia and thrombocytopenia, arthritis, exanthema, and polyserositis, to lasting isolated damage to the kidneys, bone marrow, and joints. The disease preferentially affects childbearing age females (ratio F:M 10:1) [6]. The course of SLE is often unpredictable, with many attacks and milder forms of SLE showing spontaneous remission. Renal involvement in SLE is associated with high mortality. With aggressive therapeutic schemes, survival rates have been steadily increasing in recent years. The American Rheumatology Association compiled the seven most important diagnostic criteria for SLE [207].
SLE involves increased production of autoantibodies, immune complex deposition in the microvasculature of various organs, complement activation, leukocyte infiltration, and tissue damage. The immune complex glomerulonephritis of SLE is a major cause of morbidity and a determinant of the outcome of the disease [209]. Advances are needed in the treatment of severe lupus erythematosus, both to reduce the current mortality rate of 10– 20% after 10 years and to decrease the development of renal insufficiency requiring dialysis, which occurs in nearly one quarter of patients. In addition, efforts must continue to minimize the adverse effects of long-term immunosuppressive therapy [209]. TPE is particularly indicated in severe cases, such as:- Rapid progression despite immunosuppressive therapy, renal involvement, e.g., proliferative glomerulonephritis and nephrotic syndrome [208,210], extremely acute generalized vasculitis [208], thrombocytopenia and leukopenia, pulmonary, cardiac, and cerebral involvement, pancreatitis [49].
Prolonged treatments have been reported, but their rationale and efficacy are questionable [6]. According to Clark et al., TPE with 4 L once per month probably modulates the immune response and thus intervenes beneficially in the course of the disease [6].
Cyclosporin A is a well-known immunosuppressive drug that has been used successfully for many years to delay organ transplant
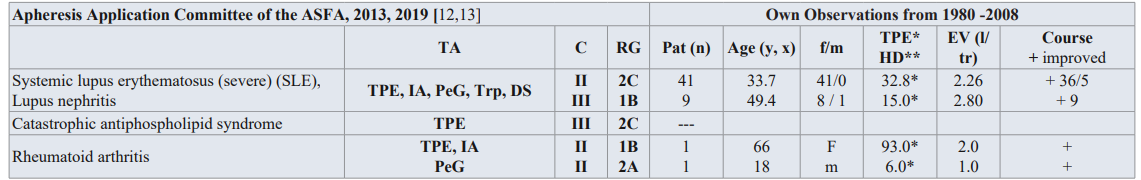
Table 5: Guidelines on the use of TA in clinical practice-based approach in autoimmune diseases with immunologic origin (12, 13), and own observation from 1980-2008.
TA: TA modality; C: Category; RG: Recommendation grade; Pat.: patient; f/m: female/male; EV: Exchange volume (l/tr: liter / treatment,x); Substitution: 3-5 % human albumin electrolyte solution, FFP; PeG: Peptid-Gam®: synthetic peptide-goat-antimouse; Trp: Tryptophan; DS: Dextran sulfate; IA: Immonadsorption on protein A.
Category I: accepted for TA as first-line therapy; Category II: accepted for TA as second-line therapy; Category III: not accepted for TA, decision should be individualized; Category IV: not accepted for TA, approval is desirable if TA is undertaken [12,13].rejection in particular. Cyclosporin A seems to be promising in the management of autoimmune diseases, and via a similar mechanism of immune suppression as observed in animal experiments and in vitro studies. Routine implementation of cyclosporin A in chronic SLE presents new therapeutic possibilities due to selective inhibition of T cell activity at a very early stage [211]. The prognosis for SLE with varying organ manifestations has been considerably improved in recent years due in part to very aggressive therapy schemes [212].
Antiphospholipid Syndrome (APS)
APS is an autoimmune hypercoaguable state caused by antibodies against cell membrane phospholipids that provoke thrombosis in the arteries and veins. Antiphospholipid antibodies can be detected by measuring lupus anticoagulant and anticardiolipin antibodies [207]. Antiphospholipid antibodies are implicated in vascular thrombosis, thrombocytopenia, and recurrent fetal loss in patients with SLE. The etiology of thrombosis of the small and large vessels is not completely understood. Involvement of the kidneys in APS is possible. In addition, of thrombosis of the great arteries and veins, microscopic thrombotic microangiopathy is typically observed on kidney histology. High levels of antiphospholipid antibodies in patients with SLE increase the risk of venous and arterial thrombosis, adverse cerebrovascular events, recurrent fetal loss, and other arterial thrombotic and embolic complications, such as superior mesenteric artery thrombosis and thrombocytopenia. While APS can exist without SLE, it should also be considered in non-SLE patients when classical symptoms, such as recurrent thrombosis of unknown etiology, are present. TA can be considered life-saving in patients with severe APS [213].
Catastrophic antiphospholipid syndrome (CAPS) is an acquired hypercoagulable state, an unusual variant of APS. CAPS is defined as the acute onset of multiple thrombosis in at least three organ systems over a period of days or weeks, in patients with serologic evidence of antiphospholipid antibodies (lupus anticoagulant, anticardiolipin antibodies, and/or anti-β2 glycoprotein I). The most commonly affected sites are small vessels of kidneys, lungs, brain, heart, and skin, although large vessel thrombosis can also be present [12].
The exact mechanism by which TPE exerts an effect in CAPS is not known, but removal of pathologic antiphospholipid antibodies, as well as cytokines, tumor necrosis factor-α (TNF-α), and complement, is thought to play an important role. In most reports in which the replacement fluid transfusion of natural anticoagulants such as protein C, protein S, and antithrombin is given, this may contribute to the overall benefit of this procedure. However, it has not been established if plasma transfusion alone would have similar benefits because this option has not been tested. The category III for TPE is assigned based on a paucity of data (Table 5) [13].
The optimal treatment of CAPS is still debatable given that the condition is rare and there have been no relevant prospective studies. However, the therapeutic approach has to have three aims:
- to treat any precipitating factors, g., infection, organ necrosis,
- to prevent and to control ongoing thrombosis, 3) to suppress excessive cytokine production [12].
First-line therapies from retrospective study data should always include the combination of anticoagulation against thrombosis, glucocorticoids plus TA, and/or intravenous immunoglobulins in the treatment of CAPS [214]. If CAPS is associated with a flare of SLE, cyclophosphamide is also used. In combination with infection, parenteral antibiotics should be administered [12]. A minimum of 3–5 TPEs are recommended. Discontinuation is based on the patient’s clinical response. Some patients have been treated for weeks. TPE and IA are valuable treatment strategies in patients with refractory disease manifestations and in pregnancy. IA seems to have a favorable side effect spectrum compared to TPE. There is a clear need to perform randomized controlled trial to evaluate efficacy, safety and tolerability of both treatment strategies in the treatment of SLE and CAPS [215,216].
Rheumatoid Arthritis (RA)
RA is an autoimmune disease that affects approximately 1–3% of the population and results in considerable morbidity and debility [6]. A typical characteristic of RA is that the joints are affected, with accompanying extra-articular manifestations, such as vasculitis as well as spleen and lymph node involvement. Recent evidence supports a central role for activated T cells in its pathogenesis. In the inflamed joints of patients with rheumatoid arthritis, activated T lymphocytes accumulate as activated cells [217]. The etiology and pathogenesis of rheumatoid arthritis are still unclear for the most part. It is known that treatment of this disease is very difficult and even controversial. Most drugs have only limited efficacy.
In RA, which is a chronic multisystem autoimmune disease, the most characteristic feature is an inflammatory synovitis, it can be relapsing or persistent, usually involving peripheral joints in a symmetric distribution. In about 20 % of the patients, there are extra-articular features, too. The role of antibodies to cyclic citrullinated peptides in the pathogenesis and diagnosis has been increasing attention [12].
A positive rheumatoid factor can be serologically detected in about 80% of patients; antinuclear antibodies, circulating immune complexes, cryoglobulins, and hypergammaglobulins may also be present. The rheumatoid factors belong to the IgM and IgG group. The immune complexes can activate the complement system and, via subsequent activation of mononuclear and polymorphonuclear cells, cause tissue damage through release of proinflammatory cytokines, particularly TNF [207]. RA has significant systemic effects, with associated morbidity and mortality. The role of humoral versus immune activity in the resulting disease process is not completely understood. T cells are activated by an unknown initiating process, resulting in production of interleukin-1 and TNF-α, which have been shown to have a significant role in the inflammatory process. It is believed that autoantigens develop after initiation, perpetuating T cell activity and the disease process [218].
The goals of therapy for rheumatoid arthritis are relief of pain; reduction of inflammation; protection of articular structures; maintenance of function; control of systemic involvement; healing of bone erosions. None of the current therapeutic interventions is curative, and all must be viewed as palliative, aimed primarily at relieving the signs and symptoms of the disease [13]. Medical management of RA can be divided conveniently into five groups of medications: Aspirin, other nonsteroidal anti-inflammatory drugs, and simple analgesics; low-dose oral glucocorticoids; disease- modifying antirheumatic drugs (e.g., methotrexate); cytokine- neutralizing agents (i.e., antiTNF, anti-IL-1); immunosuppressive and cytotoxic drugs, and novel and effective biologic agents like rituximab (13).
Because both cellular and humoral mechanisms are involved in the pathogenesis of rheumatoid arthritis, in recent years TPE, cryofiltration, lymphoplasmapheresis, and leukocytapheresis have been implemented in addition to immunosuppressive therapy in particularly severe cases [219,220]. The clinical results of cryofiltration, double filtration, IA, and leukocytapheresis are very encouraging; these methods could be a regular therapy for rheumatoid arthritis, particularly in those patients with poorly controlled disease on immune suppressive or anti TNF therapy [221,222].
After the guidelines of the AAC of the ASFA, RA has for immunoadsorption with protein A category II, (Table 5) [13]. The rationale for using staphylococcal protein A column is that protein A has a high affinity for Fc portion of IgG and for high molecular weight IgG and IGM complexes [13]. IgG antibodies and CICs can be selectively removed from the blood by perfusion of patient plasma through the columns. The removal or alteration of CICs by IA, could be immunomodulatory and potentially beneficial for patients with RA. Only small amounts of immunoglobulin are removed by IA (1–3% of total serum Igs) and their concentration is unchanged, as are plasma levels of CICs. An indirect immunomodulatory mechanism is suggested in IA-induced therapeutic responses in RA [6]. The usual treatment course is 12 weeks. In most studies, clinical improvement was delayed for up to a few weeks after completing the procedures.
The current management and treatment of rheumatoid arthritis is first to use the above mentioned five groups of medications with aspirin and other nonsteroidal anti-inflammatory drugs, and lastly, immunosuppressive and cytotoxic drugs. The biological agents can be used to target specific cells and cytokines. These drugs have been shown to reduce inflammation significantly and to retard the progression of joint damage in rheumatoid arthritis, thereby reducing symptoms and improving function [223]. Early clinical results of monotherapy using tocilizumab, anti-interleukin-6 receptor antibody, in rheumatoid arthritis were excellent [224]. Therefore, TA is only indicated in severe cases of rheumatoid arthritis if all five groups of drugs have failed. The excellent results mentioned previously may be one reason why production of Staphylococcal protein A agarose (Immunosorba) and Staphylococcal A silica (Prosorba; both Fresenius HemoCare GmbH, Germany) columns.
Inflammatory Eye Disease
When conventional therapy with cortisone or immunosuppressive drugs fails or is inadequate in the treatment of immune-mediated inflammatory eye disease with an auto immunologic pathogenesis, TA may be indicated and is increasingly being implemented with success.
Severe uveitis is potentially associated with visual impairment or blindness in young patients [225]. In posterior uveitis, progredient inflammatory processes can lead to morphologic changes in the chorioidea and retina, contributing to functional deterioration. In uveitis intermedia, inflammatory processes in the peripheral retina and in the area of the ciliary body require primary attention and aggressive treatment. In both cases, secondary destructive changes in the vessels can occur, causing reduced perfusion of the retina and chorioidea. Primary inflammatory vascular changes may lead to secondary morphologic chorioretinal changes, which may then further impair function. The inflammatory process and/or the reduced chorioretinal perfusion are important. Therefore, an anti-inflammatory/ immunomodulatory therapy, a hemorheologic therapy, or a combination of both treatments, should bring about improvement of the condition, insofar as no other specific therapy is indicated [207].
Detection of immune complexes or autoantibodies in uveitis is problematic. First, indications for the existence and possible pathomechanism of pathogenic substrates to retinal S antigen were found in patients with uveitis and in animal studies. Both improvement and deterioration in the condition can be regarded as an indication of elimination of a pathogenic substrate.
The improvement in hemorheologic parameters could contribute considerably to the therapeutic success in autoimmune eye diseases accompanied by primary or secondary vascular changes. With improved microcirculation, the damaged tissue can recover. In addition, other mechanisms, such as elimination of a pathogenic substrate or immunomodulatory effects of the exchange medium, probably contribute to the success of this therapy. The immunomodulating mechanism of TA, which favors a prompter elimination of inflammation, increases ocular function, and reduces recurrence, has been clarified.
In recent years, the anti-TNF-α antibodies, infliximab and adalimumab, and others demonstrated significant efficacy in controlling uveitis associated with seronegative spondyloarthropathies and juvenile idiopathic arthritis [226]. The majority of reports of biologic therapies in posterior uveitis have been uncontrolled or retrospective studies in patients with uveitis resistant to immunosuppression. Biologic therapies have increased the treatment options for sight-threatening uveitis. Additionally, the high cost and potential side effects of the biologic agents have limited their current use to uveitis refractory to immunosuppression. Further controlled randomized multicenter studies of TPE and/ or immunosuppression versus biologics are necessary to clarify efficacy, side effects, and costs.
Summary
Different immunological diseases can be treated by various apheresis methods. However, there are only a few prospective controlled trials available to allow definitive conclusions. RPGN is a clinico-pathologic entity consisting of rapid loss of renal function, usually a 50 % decline in GFR within some months. Therefore, TA is indicated in RPGN (ANCA associated) with dialysis dependence (Cr > 6 mg/dL), and in RPGN with diffuse alveolar hemorrhage (anti-glomerular basement membrane disease). TA in RPGN with dialysis independence is only indicated in severe cases if the immunosuppressive therapy has failed. In approximately 60 % of patients with RPGN present with crescentic glomerulonephritis (pauci-immune RPGN) with few or absent deposits, some trials have evaluated the efficacy of TA as an adjunct to conventional immunosuppressive therapy. FSGS is caused by a variety of factors, however, one type that recurs after transplantation and has been with circulating factors, can be treated with TA. MPGN from cryoglobulinemia could be an indication for TA, too. The rational for TA in HUS is discussed controversially. The treatment strategy is dependent on disease severity. TA and biologic agents, such as eculizumab, in combination seems to be prudent. TA is indicated in renal transplantation in ABO compatible antibody mediated rejection, desensitization, living donor, and positive crossmatch due to donor specific HLA antibody. In renal transplantation, ABO incompatible, TA is indicated for desensitization live donors and in humoral rejection.
TA, besides corticosteroids, IVIG, and immunosuppressive drugs, has been established as first-line therapy in a large number of neurological diseases. Especially immune-mediated neurological and hematological diseases that without treatment can lead to significant disability and in a limited number of patients to death [6]. However, a specific therapy for an individual patient is dictated by several factors, including patient comorbidity and the practice environment. An improved understanding of antibody responses and genetic backgrounds in immune-mediated neurological and hematological disorders may offer new opportunities for target interventions. Newer therapy modalities such the human monoclonal antibodies rituximab, eculizumab, belimumab, or others showed clinically improvement in severe and refractory immune-mediated neurological disorders [6]. Further controlled multi-center studies must show the effectivity of these human monoclonal antibodies in immune mediated disorders.
PV is a classic example of antibody-induced immune dermatosis. TPE or IA and ECP are indicated in patients with severe symptoms who either received high doses of conventional agents and/or had an aggressive and rapidly progressive disease. BP is another rare form of subepidermal blistering pemphigus. BP is not as dramatic as other autoimmune diseases with good response of conventional therapy. TPE and IA in combination with immunosuppression are indicated in BP and d-penicillamine-induced pemphigus only in severe cases. In severe cases of progressive scleroderma, dermatomyositis, TEN, psoriasis vulgaris, and HST, TPE or IA can be successful. In these diseases, which do not respond to this therapy, the first-line therapy is immunosuppression in usually dosages. Other immuno-suppressants, biologics, or TA could act as second-line therapy. TA is only indicated in severe cases of BehcÄ?t disease, PG, dermatitis herpetiformis, and herpes gestations and other autoimmune diseases as second-line therapy. In these cases, TA must be combined with an immunosuppressive therapy and/or biologic agents.
In other autoimmune diseases such as SLE RA, and inflammatory eye diseases, the indication of TA is controversially discussed. Only in severe refractory autoimmune diseases, TA is indicated. The use of never technologies, such as IA in combination with recent biologics, might some offer some new perspectives for extracorporeal treatment of systemic autoimmune diseases.
However, all mentioned therapeutic apheresis methods are still technically complicated and very expensive. A reduction in costs is a valid demand in view of the scarce resources available in the healthcare system. Commissions consisting of physicians, administration specialists and representatives of the health insurance funds and others nowadays decide at a “round table” who will be granted medical facilities and who will not; this is a clinical routine adopted in German. Physicians are committed to helping all patients entrusted to them to the best of their knowledge, and this means that medical treatment – and particularly the apheresis processes – must become affordable. This demand represents a great challenge to physicians, politicians, health organizations, and above all to the manufacturers. Industry constantly justifies the high costs with the extensive research and development required. All those involved in the health-care system must intensify their cooperation in this respect.
However, for all mentioned diseases the quotient relevant for cost-effectiveness assessment (cost of treatment – cost saved): (improvement in life quality) must be discussed and calculated exactly by all involved persons. After Malchesky, every effort should be made to delay the progression of acute and chronic diseases. TA is clearly an important tool in treatment of many complex conditions now and in the future [227].
References
- Afazali M, Oveisgharan S, Rajabkhah S, et Complications of therapeutic plasma exchange in patients with neurologic disorders. Curr J neurol. 2020; 19: 18-22.
- Bambauer R, Latza R, Bambauer C, et al. Therapeutic apheresis in autoimmune Rheumatol Res Rev. 2013; 5: 93-103.
- Wallukat G, Reinke P, Dörffel WW, et al. Removal of autobodies in dilated cardiopathy by immu-noadsorption. Int J Cardiol. 1996; 54: 191-195. https://www.ncbi.nlm.nih.gov/pubmed/8803685 .
- Hiepe F, Pfüller B, Wolbart K, et C1q: multifunctional ligand for a new immunoadsorption treatment. Ther Apher. 1999; 3: 246-251: https://www.ncbi.nlm.nih.gov/pubmed/10427623.
- Kumlien G, Ullsröm L, Losvall A, et al. Clinical experience with a new apheresis filter has specifi-cally depletes ABO blood group antibodies. Transfusion. 2006; 46: 1568-1575.
- Bambauer R, Latza R, Burgard D, et Therapeutic Apheresis in Hematologic, Autoimmune and Dermatologic Diseases with Immunologic Origin. Ther Apher Dial. 2016; 20: 433-452.
- Hafer C, Golla P, Gericke M, et al. Membrane versus centrifuge-based therapeutic plasma exchange: a randomized prospective crossover Int Urol Nephrol. 2016; 48: 133-138.
- Malchesky PS. Membrane processes for plasma separation and plasma fractionation. Guideline Prin-ciples for clinical use. Ther Apher. 2001; 5: 270-282.
- Kaplan AA. Why nephrologists should perform therapeutic plasma exchange. Dial Transplant. 2009; 38: 65-70.
- Madoer F, Lazarus JM, Brady HR, et al. Therapeutic plasma exchange in renal disease. J Am Soc Nephrol. 1996; 7: 367-
- Bambauer R, Bambauer C, Latza R, et al. Therapeutic apheresis in nephrology. Clin Nephrol Urol Sci. 2014. Doi: 72243/2954-7161-1-2
- Schwartz J, Winters JL, Padmanabhan A, et al. T Guidelines on the Use of Therapeutic Apheresis in Clinical Practice– Evidence-Based Approach from the Writing Committee of the American Society for Apheresis: The Sixth Special J Clin Apher. 2013; 28: 145-284.
- Padmanabhan A, Connelly-Smith L, Aqui N, et Guidelines on the Use of Therapeutic Aphere-sis in Clinical Practice – Evidence-Based Approach from the Writing Committee of the American Society for Apheresis: The Eighth Special Issue. J Clin Apher. 2019; 34: 171-354.
- Bambauer R, Schiel R, Lehmann B, et al. Therapeutic Apheresis. Technical ARPN J Sci Technol. 2012; 2: 339-421.
- Davidson A, Diamond B. Autoimmune diseases. N Engl J Med. 2001; 345: 340-350.
- Bambauer R, Latza R, Burgard D, et al. Auto-Immune Disorders treated with Therapeutic Immunol R Tes Ther J. 2017; 1: 111.
- Bambauer R, Latza R, Schiel R. Therapeutic Apheresis in Immunologic Renal and Neurologic Dis-eases. Ther Apher Dial. 2017; 21: 6-21.
- Bambauer Therapeutic Apheresis and Immunosuppression in Renal Diseases. J Clin Res Reports. 2021; 7: 1-15.
- Stummvoll G, Aringer M, Handisurya A, et al. Immunoadsorption in Autoimmune Diseases Affecting the Kidney. Sem Nephrol. 2017; 37: 478-487.
- Walsh M, Merkel PA, Peh CA, et al. CA. Plasma exchange and glucocorticoid dosing in the treatment of antineutrophil cytoplasm antibody associated vasculitis (PEXIVAS): protocol. Trials. 2013; 14: 73.
- Balogun RA, Sanchez AP, Klingel R, et al. Update to the ASFA guidelines on the use of therapeu-tic apheresis in ANCA-associated J Clin Apher. 2020; 35: 493-499.
- Walsh M, Merkel PA, Peh CA, et al. Plasma Exchange and Glucocorticoids in severe ANCA-Associated Vasculitis. N Engl J Med. 2020; 382: 622-631.
- Lin J, Markowitz GS, Valeri AM, et al. Renal monoclonal immunoglobulin deposition disease. The disease spectrum. Am Soc Nephrol. 2001; 12: 1482-1492.
- De Lind van Wijngaarden RA, Hauer HA, Wolterbeck R, et al. Clinical and histologic determinants of renal outcome in ANCA-associated vasculitis: A prospective analysis of 10 patients with severe renal involvement. J Am Soc Nephrol. 2006; 17: 2264-2272.
- De Lind van Wijngaarden RA, Hauer HA, Wolterbeck R, et for the European Vasculitis Study Group (EUVAS). Changes of renal recovery for dialysis-dependent ANCA associated glomerulone-phritis. J Am Soc Nephrol. 2007; 18: 2189-2189.
- Prendecki M, Pusey C. Plasma exchange in antiglomerular basement membrane Presse Médicale. 2019; 48: 328-337.
- Yamada Y, Harada M, Hara Y, et al. Efficacy of plasma exchange for antineutrophil cytoplasmic antibody associated systemic vasculitis: a systematic review and meta-analysis. Arch Res Ther. 2012; 23: 141.
- Guo S, Mühlfeld AS, Wietecha TA, et Deletion of acting Fcγ receptors does not confer protection in murine cryoglobulinemia-associated membranoproliferative glomerulonephritis. Am J Pathol. 2009; 175: 107-116.
- Clynes R, Dumitru C, Ravetch JV. Uncouplin of immune complex formation and kidney damage in autoimmune glomerulonephritis. Science. 1983; 279: 1052-1054.
- Lockwood CM, Pusey CD, Peters Indications for plasma exchange: Renal disease. In: Lysaght MJ, Gurland HJ (eds.) Plasmaseparation and plasma Fractionation. Karger AG, Basel, Switzerland. 1983; 145-152.
- Tensaki M, Takashi H, Sato R, et al. Successful Treatment with Multitarget Therapy of myco-phenmolate Mofetil and Takrolimus for Cyclophosphamide-resistant Antineutrophil Cytoplasmic Developed Independently of Systemic Lupus Erythemathosus. Clin Rheumatol. 2021; 27: 79-80.
- Jayne DRW, Gaskin G, Rasmussen N, et al. European Vasculitis Study Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vascu-litis. J Am Soc Nephrol. 2007; 18: 2188- 2188.
- Hasegawa M, Kawamura N, Kasugai M, et Cytapheresis for the treatment of myeloperoxidase antineutrophil cytoplasmatic anti-associated vasculitis: Report of 5 cases. Ther Apher. 2007; 6: 443- 449.
- Ewert BH, Jennette JC, Falk RJ. Anti-myeloperoxidase antibodies stimulate neutrophils to damage human endothelial cells. Kid Int. 1992; 41: 375-383.
- Cole E, Caffran D, Magli A, et al. A prospective randomized trial of plasma exchange as additive therapy in idiopathic crescentic The Canadian Apheresis Study Group. Am J Kid Dis. 1992; 20: 261-270.
- Cascian AL, Jayne DRW. Role of plasma exchange in the treatment of primary vasculitis. Int J Clin Rheumatol. 2010; 5: 339-344.
- Hayes JS, Chang J, Abdel-Rahman EM, et al. Therapeutic plasma exchange for related conditions in the elderly: Ten years’ experience in one Sem Dial. 2012; 25: 159-164.
- Walsh M, Catapano F, Szpirt W, et Plasma exchange for renal vasculitis and idiopathic rapidly progressive glomerulonephritis: A meta-analysis. Am J Kid Dis. 2011; 57: 566-574.
- Tang Al, Davies DR, Wing WJ. Remission of nephrotic syndrome in amyloids associated either a hypernephroma. Clin Nephrol. 1989; 32: 225-235.
- Bagga A, Hari P, Moudgil A. Mycophenolate mofetil and prednisolone therapy in children with steroid-dependent nephrotic syndrome. Am J Kid Dis. 2003; 42: 1114-1120.
- Bagga A, Mantan M. Nephrotic syndrome in children. Ind J Med Red. 2005; 120: 13-19.
- Habashy D, Hodson EM, Craig JC. Interventions for steroid- resistant nephritic syndrome: a system-ic review. Pediatr Nephrol. 2004; 18: 906-912.
- Kashtan C, Melvin T, Kim Long-term follow-up of patients with steroid-dependent minimal change glomerulonephritis. Clin Nephrol. 1988; 29: 79-85.
- Ronco PM, Alyanakian MA, Mougenot B, et al. legit chain deposition disease: A model of glomerulo-sclerosis defined at the molecular level. Am Soc Nephrol. 2001; 12: 1558-1565.
- Matto TK, Mahmoud MA. Increased maintenance corticosteroids during upper respiratory infection decrease the risk of relapse in nephritic Nephrol. 2000; 85: 342-348.
- Nakayama M, Katafuchi R, Yanase T, et al. Steroid responsiveness and frequency of relapse in adult – onset minimal change nephrotic Am J Kid Dis. 2002; 39: 503-512.
- Musco E, Mune M, Fujii Y, et al. The Kansai FGS LDL- apheresis-FLAT Study Group. Nephron. 2001; 89: 408-414.
- Alshaya HO, Al Maghrabi JA, Kari JA. Intravenous pulse cyclophosphamide – is it effective in chil-dren with steroid- resistant nephrotic syndrome? Pediatr Nephrol. 2006; 18: 1143-1146.
- Dantal J, Godfrin Y, Koll R, et Antihuman immunoglobulin affinity immunoadsorption strongly decreases proteinuria in patients with relapsing nephritic syndrome. J Am Soc Nephrol. 1998; 9: 1709-1715.
- Esnault VL, Besnier D, Testa A, et Effect of protein A immunoadsorption in nephrotic syndrome of various etiologies. J Am Soc Nephrol. 1999; 20: 2014-2017.
- Russo GE, Bonello M, Bauco B, et al. Nephrotic syndrome and plasmapheresis. Int J Art Org. 1999; 23: 111-117.
- Haas M, Godfin Y, Oberbauer R, et al. Plasma immunoadsorption treatment in patients with primary focal and segmental glomerulonephritis. Nephrol Dial Transplant. 1998; 13: 2013-2017.
- Bambauer R, Latza R, Schiel Therapeutic apheresis in the treatment of hemolytic uremic syndrome due to pathophysiologic aspect. Ther Apher Dial. 2011; 15: 10-19.
- Zimmerhackl LB, Verweyen H, Gerber A, et Das hämolytisch- urämische Syndrom. Dtsch Ärztebl. 2002; 99: 197-203.
- Bläker F, Altrooge H, Hellwege HH, et Dialysebehandlung des schweren hämolytisch-urämischen Syndroms. Dtsch Med Wschr. 1978; 103: 1229-1233.
- Spencer CD, Crane FM, Kumar JR, et al. Treatment of postpartum haemolytic uremic syndrome with plasma exchange. JAMA. 1982; 247: 2808-2809.
- Gillor A, Roth B, Bulla M, et al. Plasmapherese bei der Behandlung von Kindern mit hämolytisch-urämischen Nieren- Hochdruckkrankh. 1986; 15: 118-123.
- Bambauer R, Jutzler GA, Jesberger HJ, et Therapeutischer Plasma-Austausch beim hämolytisch-urämischen Syndrom. Dtsch Med Wschr. 1988; 113: 1245-1249.
- Dittrich E, Schmaldienst S, Derfler Plasmaaustausch und Immunadsorption. Wien Klin Wschr. 2007; 2: 39-54.
- Kaplan AA. Therapeutic apheresis for cancer related uremic syndrome. Ther Apher. 2001; 4: 201- 206.
- Michael M, Elliot EJ, Ridley GF, et Interventions for haemolytic uraemia syndrome and throm-botic thrombocytopenic purpura. Cochrane Database Sys Rev. 2009; 53: 259-272.
- O´Regan S, Blais N, Russo P, et al. Hemolytic uremic syndrome: glomerular filtration rate, 6 to 11 years later measured by 99mTc DTPA plasma slope clearance. Clin Nephrol. 1989; 32: 217-220.
- Rasco DA, Webster DR, Sahl JW, et Origins of the E. coli strain causing outbtreak of haemolytic-uremic syndrome in Germany. N Engl J Med. 2011; 365: 709-717.
- Kielstein JT, Beutel G, Feig S, et Best supportive care and therapeutic plasma exchange with or without eculizumab in Shiga-toxin-producing E. coli O104:H4 induced haemolytic uremic syndrome: an analysis of the German STEC-Hus registry. Nephrol Dial Transplant. 2012; 27: 1807-1815.
- Teschner S, Kurschat C, Burst V. Therapeutic apheresis in transplantation: overview and critical evaluation of available modalities in respect to indications evidence and costs. Transplanta-tionsmedizin. 2010; 22: 266-272.
- Mao Y, Bai J, Chen J, et A pilot study of GC/MS- based serum metallic profiling of acute rejection in renal transplantation. Transplant Immunol. 2008; 19: 74-79.
- Gloor JM, Winters JL, Cornell LD, et al. Baseline donorspecific antibody levels and outcomes in positive cross-match kidney transplantation. Am J Transplant. 2010; 10: 582-588.
- Ichimaru N, Takahara Japan´s experience with living-donor kidney transplantation across ABO barriers. Nat Clin Pract Nephrol. 2008; 4: 682-692.
- Sawada T, Fuchinoue S, Teraoka Successful A1-to O ABO- incompatibility kidney transplantation after a preconditioning regimen consisting of anti-CD20 monoclonal antibody infusions, splenecto-my, and double filtration. Transplantation. 2009; 74: 1207-1214.
- Donauer J, Wilpert J, Geyer M, et al. ABO-incompatible kidney transplantation using antigen-specific immunoadsorption and rituximab: a single center experience. Xenotransplantation. 2006; 13: 108-114.
- Scharma A, Bummerts J, Gomez-Navarro D, et al. Clearance for monoclonal antibody (mab) CP-675,206 by therapeutic plasma exchange (TPE) or plasmapheresis. J Clin Oncol. 2007; 25: 1515-1519.
- Franz HJC, Rahmel A, Doxiadis II. Enhanced kidney allocation to highly sensitized patients by an acceptable mismatch program. Transplantation. 2009; 88: 447-452.
- Gloor JM, DeGoey SR, Pineda AA, et al. Overcoming a positive cross-match in liver donor kidney transplantation. Am J Transplant. 2003; 3: 1017-1022.
- Jordan SC, Tyan D, Stablein D, et Evaluation of intravenous immunoglobulin as an agent to lower allosensitization and improve transplantation in highly sensitized adult patients with end-stage renal disease: report of the NIH IG02 trial. J Am Soc Nephrol. 2004; 15: 3256-3262.
- Ashley A Vo, Marina Lukovsky, Mieko Toyoda, et al. Rituximab and intravenous immune globulin for desensitiza- tion during renal transplantation. N Engl J Med. 2008; 359: 242-248.
- Venetz JP, Pascual M. New treatments for acute humoral rejection of kidney Inform Healthca. 2007; 16: 625-631.
- Higgins R, Zehnder D, Chen J, et al. The histological development of acute antibody-mediated rejection in HLA antibody-incompatible renal transplantation. Nephrol Dial Transplant. 2010; 25: 1306-1312.
- Takemoto SK, Zevi A, Feng S, et National conference to assess antibody-mediated rejection in solid organ transplantation. Am J Transplant. 2004; 4: 1033-1141.
- Böhmig GA, Wahrmann M, Regele H, et Immunoadsorption in severe C4d-positive acute kidney allograft rejection: a randomized controlled trial. Am J Transplant. 2001; 7: 117-122.
- Bell WR, Braine HG, Ness PM, et al. Improved survival in thrombotic thrombocytopenic purpura syndrome: Clinical experience in 108 N Engl J Med. 1991; 325: 398-405.
- Byrne MC, Budisavijevic MN, Fan Z, et al. Renal transplant in patients with Alport´s syndrome. Am J Kid Dis. 2002; 39: 769-773.
- Laczika K, Knapp S, Derfler K, et Immunoadsorption in Goodpasture´s syndrome. Am J Kid Dis. 2000; 36: 392-397.
- Bolton Goodpasture´s syndrome. Kid Int. 1996; 50:1753-1759.
- Dantal J, Bigot E, Bogers W, et al. Effect of plasma protein adsorption on protein excretion in kidney-transplant recipients with recurrent nephritic N Engl J Med. 1994; 330: 7-14.
- DallAmico R, Ghiggeri G, Carraro M, et al. Prediction and treatment of recurrent focal segmental glomerulosclerosis after renal transplantation in children. Am J Kid Dis. 1999; 34: 1048-1055.
- Bambauer R. Therapeutic Apheresis in Renal Diseases with Immunologic Origin. Ann Immunol Immunother.2020; 2:
- Shelat Practical considerations for planning a therapeutic apheresis procedure. Am J Med. 2010; 123: 777-784.
- Ariga T, Yu RK. Antiglycolipid antibodies in Guillain-Barré syndrome and related Review of clinical features and antibody specifities. J Neurosci Res. 2005; 80: 1-17.
- Miller AC, Rashid RM, Sinert Guillain-Barré syndrome. EMedicine specialities >Emergency EMedicine> Neurolgy update Apr. 23. 2010). 792008-overview.
- Mancardi GL, del Sette M, Primavera A, et al. Borrelia burgdorferi infection and Guillain-Barré syndrome. Lancet. 1989; 21: 985-986.
- Hartung KP, Heininger K, Toyka KV. Guillain-Barré Syndrome: Prognoses und Lancet. 1989; I: 109.
- Koo YS, Shin HY, Kim JK, et al. Early Electro diagnostic Feature of Upper Extremity Sensory Nerves Can Differentiate Axonal Guillain-Barre Syndrome from Acute Inflammatory Demyelinating J Clin Neurol. 2016; 12: 495-501.
- Davis AJ, Fehmi J, Senec M, et al. Immunoadsorption and Plasma Exchange in Seronegative Immune-Mediated Neuropathies. J Clin Med. 2020; 9.
- Bambauer R, Schiel R. Therapeutic Apheresis in Neurology. J Clin Res Rep. 2020.
- Korintenberg R, Schessl J, Kirchner J, et al. Intravenous administered immunoglobulin in the treatment of childhood Guillain-Barré-syndrome: A randomized trial. Pediatrics. 2005; 116: 8-14.
- R Weinstein. Therapeutic apheresis in neurologic disorders. Ther Apher. 1999; 3: 161-171.
- Dada MA, Kaplan Plasmapheresis treatment in Guillain- Barré syndrome. potential benefit over IVIG in patients with axonal involvement. Ther Apher Dial. 2004; 8: 409-412.
- Haupt WF, Birkmann C, van der Ven G, et Apheresis and selective adsorption plus immuno-globulin treatment in Guillain-Barré syndrome. Ther Apher. 2000; 4: 198-200.
- Ramdhani VM, Suwarman K, Sudjud RE, et Plasmapheresis in patient with Guillain-Barré syndrome of the intensive care unit. J Health Med Nurse. 2020; 5: 13-24.
- Pelletier JPR, Mukhtar F. Chapüter 16-Passive Monoclonal and Polyclonal Antibody Therapies. Immunol Conc Transf Med. 2020; 251-348.
- Vendramin C, Scully Indications of plasma exchange in combination with intravenous immunoglobulins or therapeutic monoclonal antibodies. How to combine them? La Presse Medicale. 2019; 48: 354-359.
- Dyck PJB, Iracy History, diagnosis, and management of chronic inflammatory demyelinating polyneuropathy. Mayo Clin Proc. 2018; 93: 777-793.
- Ryan M, Ryan SJ. Chronic Inflammatory Demyelinating Polyneuropathy: Considerations for Diagnosis, Management, and Population Health. Am J Manag Care. 2018; 24: 371-
- Hughes Management of chronic inflammatory demyelinating polyradiculoneuropathy. Drugs. 2003; 63:275-287.
- Hughes RAC, Bouche P, Cornblatt DR, et al. European Federation of Neurological Socie-ties/Peripheral Nerve Society guideline on management of chronic inflammatory demyelinating poly-radiculoneuropathy: report of a joint task force of the European of Neurological Societies and the Pe- ripheral nerve Europ J Neurol. 2006; 13: 326-334.
- Oji S, Nomura K. Immunoadsorption in neurological disorders. Transf Apher Sci. 2017; 56: 671-676.
- Mangotti P, Pesavento V, Buoite Stella A, et Miller Fisher syndrome diagnosis and treatment in a patient with SARS- CoV-2. J Neurovirol. 2020; 11: 1-2.
- Zhao H, Shen D, Zhou H, et al. Guillain-Barré syndrome associated with SARS-CoV-2. Lancet Neurol. 2020; 19: 383-384.
- Behin A, Le Panse R. New Pathways and Therapeutic Targets in Autoimmune Myasthenia Gravis. J Neurolmusc Dis. 2018; 5: 265-277.
- Lin W, Burgess RW, Dominguez B, et Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature. 2001; 410: 1057-1064.
- Zanhg B, Tzartos JS, Belmiezi M, et Autoantibodies to lipoprotein-related protein 4 in pa-tients with double- seronegative myasthenia gravis. Arch Neurol. 2012; 69: 445-451.
- Schneider-Gold C, Toyka KV. Myasthenia gravis: Pathogenesis and Immunotherapy. Dtsch Ärztebl. 2007; 104: 420-426.
- Bachmann K, Burkhardt D, Schreitert J, et al. Thymectomy is more effective than conservative treatment for myasthenia gravis regarding outcome and clinical Surgery. 2009; 145: 392-396.
- Miller RG, Milner-Brown HS, Dau PC. Antibody-negative acquired myasthenia gravis: Success-ful therapy with plasma exchange. Musc Nerve. 1981; 4: 255-260.
- Dogra A, Rana K, Rathod C, et al. Outcome of therapeutic plasma exchange in Myasthenia gravis patients. J Family Med Prim Care. 2020; 9: 5971-5975.
- Howard JF. Myasthenia gravis: the role of complement at the neuromuscular Ann NY Acad Sci. 2017; 1412: 113-128.
- Mantegazza R, Antozzi When myasthenia gravis is deemed refractory: clinical signposts and treatment strategies. The Adv Neurol Dis. 2018; 11: 1018.
- Vershuuren JGM, Wirtz PW, Titulaer MJ, et al. Available treatment options for the management of Lambert-Eaton myasthenic syndrome. Exp Opin Pharmacothera. 2006; 7: 1323-1336.
- Huang K, Luo YB, Yang H. Autoimmune Channelopathies at Neuromuscular Junction. Front Neurol. 2019; 10: 516.
- Motomura M, Hamasaki S, Nakane S, et al. Apheresis treatment in Lambert-Eaton myasthenic syndrome. Ther Apher. 2004; 4: 287-290.
- Shen P, Fillatreau S. Anmtibody-independent functions of B cells: a focus on cytokines. Nat Rev Immuno. 2015; 15: 441-451.
- Li RT, Rezk A, Healy LM, et al. Cytokine-defined B cell responses as therapeutic targets in multiple sclerosis. Front Immunol. 2016; 6: 110.
- Khatri BO. Therapeutic apheresis in multiple sclerosis and other central nervous system disorders. Ther Apher. 2000; 4: 263-270.
- Magana SM, Pittcock SJ, Lennon VA, et NMO-IgG status in fulminant inflammatory CNS demyelinating disorders. Arch Neurol. 2009; 66: 964-966.
- Dorst J, Fangaerau Taranu D, et al. Safety and efficacy of immunoadsorption versus plasma exchange in steroid- refractory relapse of multiple Sclerosis and clinically isolated syndrome: A randomized, parallel-group, controlled trial. Lancet. 2019; 16: 98-106.
- Mulero P, Midaglia L, Montalben X. Ocrelizumanb: a new milestone in multiple sclerosis therapy. Ther Adv Neurol Dis. 2018; 11: 1018.
- Giedratiene N, Kaubrys G, Kizlaitiene, et al. Therapeutic Plasma Exchange in Multiple Sclerosis Patients with Abolished Interferon beta Bioavailability. Med Sci Monit. 2015; 21: 1512-1519.
- Bambauer R, Schiel R. Therapeutic Apheresis in Pediatrics with Neurological and Hematologi-cal Diseases. J Clin Exp Immunol. 2020; 5: 156-172.
- Kiprov DD, Dau PC, Morand The effect of plasmapheresis and drug immunosuppression on T cell subsets as defined by monoclonal antibodies. J Clin Apher. 1983; 1: 57-63.
- Latimer ME, L´Etoile N, Seidlitz J, et Therapeutic Plasma Apheresis as a Treatment for 35 Severely III Children and Adolescents with Pediatric Autoimmune Neuropsychiatric Disorders Associated with Streptococcal Infections. J Child Adolesc Psychopharmac. 2015; 25: 70-72.
- Bien CG, Bien Autoimmune encephalitis in children and adolescents. Neurol Res Pract. 2020; 2.
- Castellano JF, Meyer JA, Lado FA. A Case Serious of Adult-Onset Rasmussen´s Encephalitis: Diagnostic and Therapeutic Challenges. Front Neurol. 2017; 8: 564.
- Moussa T, Baker L, Chehab A, et Plasma Exchange for Autoimmune Encephalitis and Demyelinating Encephalomyelitis: Diagnostic and Therapeutic Challenges. J Neuroinfect Dis. 2017; 8: 262.
- Churchwell KB, Mc Manus ML, Kent P, et al. Intensive blood and plasma exchange for treatment of coagulopathy in meningococcemia. J Clin Apher. 1995; 10: 171-177.
- Sato T, Nakaqo T, Baba M, et al. Immunoadsorption plasmapheresis in acute ataxic neuropathy. Ther Apher. 1998; 2: 71-73.
- Yammamoto T, Geigo K, Suzuki A, et Long- term improvement of idiopathic hypertrophic cranial pachymeningitis by lymphocytapheresis. Ther Apher. 2000; 4: 313-316.
- Bambauer R, Jutzler GA, Philippi H, et al. Hemofiltration and plasmapheresis in premature and Art Organs. 1988; 12: 20-26.
- Filbes D, Hanson, Wesström G. The prevalence of red cell antibodies in pregnancy correlated to the outcome of the newborn. A 12-year study in central Sweden. Acta Obstet Gynecol Scan. 1995; 74: 687-692.
- Bambauer R, Latza R, Schiel R. Therapeutic Apheresis in Neurological and Hematological Diseases in Pediatrics. J Clin Exp immunol. 2020; 5: 153-172.
- Bing JI, Ping HE, Liu H. Clinical study of 44 cases of Rh- negative pregnant women. Cin J Birth Health & Heredity. 2006; 4: 8-14.
- Yohei M, Ushijima J, Furukawa S, et Plasmapheresis for the treatment of anti-alloimmunization in pregnancy. Case Rep Obstet Gynecol. 2020.
- Wind M, Gaasbeek AGA, Oosten LEM, et al. Therapeutic plasma exchange in pregnancy: A literature review. Euro J Obstet Gynecol Reprod Biol. 2021; 260: 29-36.
- Engelfried CP, von Overbeck MAM. AEGKR die Borne Autoimmune hemolytic anemia. Sem Hematol. 1992; 29: 3-12.
- Bass GF, Tuscano ET, Tuscano JM. Diagnosis and classification of autoimmune hemolytic anemia. In: Schoenfeld Y, ME Gershwin (eds.) Diagnostic criteria in Autoimmune Autoimmun Rev. 2014; 13: 560-564.
- Sawada T, Fuchinoue S, Teraoka Successful A1-to O ABO incompatibility kidney transplan-tation after a preconditioning regimen consisting of anti-CD20 monoclonal antibody infusions, splenectomy, and double filtration. Transplantation. 2009; 74: 1207-1214.
- Semple JW, Freedman J. Autoimmune pathogenesis and autoimmune haemolytic anemia. Sem Hematol. 2005; 42: 122-130.
- King KE, Ness PM. Treatment of autoimmune haemolytic anemia. Sem Hematol. 2005; 42: 131-136.
- Hoffmann R, BenzEJJ, Schattil SJ, et al. Semin Hematol eds. Hematology: Basic Principles and Practice, 4th edn. Philadelphia: Elsevier, Churchill, Living-stone. 2005; 429-438.
- Verholen F, Stadler M, Helg C, et Resistant pure red cell aplasia after allogenic stem cell transplantation with major ABO mismatch treated by escalating dose donor leukocyte infusion. Eur J Haematol. 2004; 73: 441-446.
- Matyr S, Balakumuran A, Montero A, et al. A pilot study of rituximab in patients with moderate aplastic black fan anemia and pure red cell aplasia. Blood. 2013; 122: 2479-
- Drew MJ. Therapeutic plasma exchange in hematologic diseases and In: McLeod BC, Price H,Weinstein R, eds. Apheresis: Principles and Practice, 2nd edn. Bethesda: AABB Press. 2003; 345-354.
- Handisurya A, Worel N, Rabitsch W, et al. Antigen- specific immunoadsorption with the Glyco-sorb® ABO immunoadsorption system as a novel treatment modality in pure red cell aplasia follow-ing major and bidirectional ABO-incompatible allogenic hematopoietic stem cell Transplantation. Front Med. 2020; 7: 585-628.
- Henig I, Yehdai-Ofir D, Zoher Y, et Pure red cell aplasia following ABO-mismatched allogenic hematopoietic stem cell transplantation: Resolution with Daratummab treat- ment. Acta Haematol. 2021; 1-5.
- Drexler B, Buser A, Infanti L, et al. Extracorporeal Photopheresis in Graft-versus-Host Disease. Transf Med Hemother. 2020; 47: 214-224.
- Kremer Hovinga JA, Coppo P, Lämnmle B, et Thrombotic thrombocytopenia purpura. Nat Tev Dis Primers. 2017; 3: 17020.
- Bambauer R, Bambauer C, Schiel R. Therapeutic apheresis (TA) on thrombocytopenia. Thrombocytopenia. In: Maysaa El Sayed Zaki (ed.). Nova Science Publishers. 2012; 197-
- Izak M, Bussel JB. Management of thrombocytopenia. F 1000. Prime Rep. 2014; 6: 45.
- Scully M, Goodship T. How I treat thrombotic thrombocytopenic purpura and atypical hemolyt uremic syndrome. Br Haematol. 2014; 164: 759-766.
- Swisher KK, Terrell DR, Vesely SK, et Clinical outcomes after platelet transfusions in pa-tients with thrombotic thrombocytopenic purpura. Transfusion. 2009; 49: 873-887.
- Elessa D, Talbot A, Lambion N, et Development of thrombotic thrombocytopenic purpuraduring during lenalidomide therapy: three new cases and review of literature. Br J Haematol. 2020; 188: 332-340.
- Bambauer R, Schiel R. Thrombocytopenia treated with therapeutic apheresis, and/or Human monoclonal antibodies, and/or thrombopoietin receptor agonists. Clin Rev Cases. 2020; 2: 1-7.
- Psaila B, Bussel JB. Immune thrombocytopenic purpura. Hemat/Oncol Clin N Am. 2007; 21: 743-759.
- McMillan The pathogenesis of chronic immune (idiopathic) thrombocytopenia purpura. Sem Hematol. 2000; 37: 5-9.
- Kiefel V. Platelet antibodies and diagnosis of immune- mediated thrombocytopenia. Int Ther Transfus Med. 2001; 28: 209-216.
- Richard H Aster RH. Drug-induced immune thrombocytopenia. Pathogenesis, Diagnosis and J Thromb Haemost. 2009; 7: 911-918.
- George N, Woodson RD, Kiss E, et al. Rituximab therapy for thrombotic thrombocytopenic purpura: A proposed study of the Transfusion Medicine/Hemotasis Clinical Trials Network with a systemic review of rituximab therapy for immune mediated disorders. J Clin Apher. 2006; 21: 49-56.
- Gloor JM, Winters JL, Cornell LD, et al. Baseline donor specific antibody levels and outcomes in positive crossmatch kidney Am J Transplant. 2010; 10: 582-589.
- Kempton CL, Antun AG. Thrombotic Thrombocytopenic Purpura In: Transfusion Medicine and Third Edition. Clinical and Laboratory Aspects. 2019; 649-654.
- West KA, Anderson DR, McAlister VC, et al. Alloimmune thrombocytopenia after organ transplantation. N Engl Med. 1999; 341: 1504-1507.
- Nillsson JM, Sundquist SB, Ljung R, et al. Suppression of secondary antibody response by intravenous immunoglobulin in a patient with hemophilia B and antibodies. Scand J Haematoll. 1983; 30: 458-464.
- Collins W, Chalmers P, Hart E, et al. Diagnosis and management of acquired coagulation inhibitors: a guideline from UKHCDO. Brit J Haematol. 2013; 162: 758-773.
- Sharathkumar A, Lilicrap D, Blancette VS, et al. Intensive exposure to factor VIII is a risk factor for inhibitor development in mild J Thromb Haemost. 2004; 1: 1228-1236.
- Freiburghaus C, Berntrop E, Gunnarson M, et al. Tolerance induction using the Malmö treatment model 1995. Haemophilia. 1999; 32: 32-39.
- Friedmann J, Bewernadette GM. Immunoadsorption of factor VIII inhibitors. Curr Opion Hematol. 2004; 11: 327.
- Pollar P. Polygenic antibodies to coagulation factors. Part one: Factor VIII and Factor IX. J Thromb Haemost. 2004; 2: 1082-1095.
- Huguet HC, Lasne D, Rothschild C, et al. Extracorporeal adsorption of anti-factor VIII alloantibodies on randomly functionalized polysterene resins. Thromb Haemost. 2004; 91: 259-266.
- Driessche TV, Chuah MK. Hemophilia Gene Therapy: Ready for Prime Time? Human Gene Therapy. 2017; 28: 1013-1023.
- Bambauer R, Burgard D, Schiel Therapeutic Apheresis in Dermatological Diseases. Clin Med Insights: Therapeutics. 2015; 7: 53-62.
- Meyersburg D, Schmitt E, Kasperkiewicz M, et al. Immunoadsorption in dermatology. Ther Apher Dial. 2012: 16: 311-320.
- Hahn-Ristic K, Rzany B, Amagai M, et al. Increased incidence of pemphigus vulgaris in Southern Europeans living in Germany compared with native Germans. J Eur Acad Dermatol Venereol. 2002; 16: 68-71.
- Rolf Bambauer, Daniel Burgard, Ralf Schiel. Dermatological Diseases Treated with Therapeutic Apheresis. Goap Dermatol. 2019; 1: 35-45.
- Langan SM, Smeeth L, Hubbard R, et Bullous pemphigoid and pemphigus vulgaris-incidence and mortality in the UK: population based on cohort study. BMJ. 2008; 337: 180.
- Bambauer R, Schiel R. Therapeutic Apheresis and/or Monoclonal Antibodies in Dermatological Diseases. J Clin Investigat Dermatol. 2021; 9: 3.
- Kumar R, Jindal A, Kaur A, et al. Therapeutic Plasma Exchange-A New Dawn in the Treat-ment of Pemphigus Vulgaris. Ind J Dermatol. 2015; 60: 419.
- Tselmin S, Julius U, Bornstein SR, et Low rate of infectious complications following immunoadsorption therapy without regular substitution of intravenous immunoglobulins. Ateroscler Supplem. 2017; 30: 278-282.
- Mazzoni A, Giampietrro C, Bianco I, et al. Extracorporeal photophersis and liver transplantation: Our experience and preliminary data. Transf Apher Sci. 2017; 56: 515-519.
- Edelson R, Wu Y, Schneiderman J. American Council on ECP (ACE): Why now? consensus con-ference J Clin Apher. 2018; 33: 464-468.
- Odamaki S, Hori Y, Nakai S, et al. A Solution Kinetics simulation method for Conventional and Selective Plasma Exchange Using a Complete Mixed Reactor Model. Ther Apher Dial. 2018; 23: 266-270.
- Ludwig RJ. Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatol. 2013; 25: 812029.
- Kolesnik M, Becker E, Reinhold D, et Treatment of severe autoimmune blistering skin diseases with combination of protein A immunoadsorption and rituximab: a protocol without initial high dose or pulse steroid medication. J Eur Acad Dermatol Venereol. 2014; 28: 771-780.
- Culton DA, Qian Y, Li N, et Advances in pemphigus and its endemic pemphigus foliaceus (Fogo selvagam) phenotype: a paradigm of human autoimmunity. J Autoimmun. 2008; 31: 311-324.
- Schmidt E, Zillikens D. Immunoadsorption in dermatology. Arch Dermatol Res. 2002; 302: 241-253.
- Hörgensen EH, Clemmensen O, Anagnostaki L. Herpes gestationis. Acta Obstet Gynecol Scan. 1987; 66: 175-177.
- Ruiz-Villaverde R, Sánchez-Cano D, Ramirez-Tortosa CL. Pemphigoid gestationes: therapeutic response to pre- and postpartum immunoglobulin therapy. Actas Dermosifiliogr. 2011; 102: 735-737.
- Harris ES, Meiselman HJ, Moriatry PM, et al. Therapeutic plasma exchange for the treatment of systemic sclerosis: A comprehensive review and J Sclerod Rel Dis. 2018; 3: 132-152.
- Wolff K, Stingl Pyoderma gangrenosum. Pyod Gangr.1989; 112: 13-28.
- Attar S, Spangehl MJ, Casey WJ, et al. Pyoderma gangrenosum complicating bilateral total knee arthroplasty. Orthopedics. 2010; 33: 441.
- Reichrath J, Bens G, Bonowitz A, et al. Treatment recommendations for pyoderma gangrenosum: an evidence based review of the literature based on more than 350 patients. J Am Acad Dermatol. 2005; 53: 273.
- Fritsch PO, Sidoroff A. Drug-induced Stevens-Johnson syndrome/toxic epidermal necrolysis. Am J Clin Dermatol. 2000; 1: 349-360.
- Kim HI, Kim SW, Park GY, et al. Causes and outcomes of Stevens-Johnson syndrome and toxic epidermal necrolysis in 82 adult patients. Kor J Intern Med. 2012; 27: 203-210.
- Paquet P, Piéard GE. New insights in toxic epidermal necrolysis (Lyell’s syndrome): Clinical con-siderations, pathophysiology and target treatments revisited. Drug Safety. 2010; 33: 189.
- Harr T, French Toxic epidermal necrolysis and Stevens- Johnson syndrome. Orpha J Rare Dis. 2010; 5: 39.
- Krajeswki A, Mazurek MJ, Mlynska-Krajewski E et al. Toxic Epidermal Necrolysis Therapy with TPE and IVIG- 10 Years of Experience of the Burns Treatment Center. J Burn Care Res. 2019; 40: 652-657.
- Liumbruno GM, Centoni PE, Molfettini P, et al. Lymphocytopheresis in the treatment of psoriasis J Clin Apher. 2006; 21: 158-164.
- Jorstad S, Bergh K, Iversen OJ, et al. Effects of cascade apheresis in patients with psoriasis and psoriatic Blood Purif. 1998; 16: 37-42.
- Sohagia AB, Guptha Gunturu S, Tong TR, et al. Henoch Schönlein Purpura - a case report and review of the Gastroenterol Res Prat. 2010: 5976487.
- Bambauer R, Latza R, Schiel R. Therapeutic Plasma exchange and Selective Plasma Separation Methods. Fundamental Technologies, Pathophysiology and Clinical Results. 4th edition. 2013.
- Kuhn A, Beissert S, Krammer PH. CD4+CD25+ regulatory T cells human lupus erythematosus. Arch Dermatol Res. 2009; 301: 71-78.
- Perez De Lama G, Maier H, Nieto E, et al. Chemokine expression precedes inflammatory cell infiltration and chemokine receptor and cytokine expression during the initiation of murine lupus nephritis. J Am Soc Nephrol. 2001; 12: 369-382.
- Kasgarin M. Lupus nephritis: lesions from the path lab. Kidney Int. 1994; 45: 928-938.
- Bambauer R, Schwarze U, Schiel R. Cyclosporine A and therapeutic plasma exchange in the treatment of severe lupus erythematosus. Art Organs. 2000; 24: 852-856.
- Pagnoux C. Plasma exchange for systemic lupus erythematosus. Transfus Apher Sci. 2007; 36: 187-193.
- Ogawa A case of catastrophic antiphospholipid syndrome treated by immunoadsorption thera-py. Jpn J Apher. 1995; 14: 75-79.
- Espinosa G, Cervera Current management of catastrophic antiphospholipid syndrome. Int J Clin Rheumatol. 2011; 6: 297-303.
- Kronbichler A, Brezina B, Quintana LF, et al. Efficacy of plasma exchange and immunoadsorption in systemic lupus erythematosus and antiphospholipid syndrome: A systemic review. Autoim-mune Rev. 2016; 15: 38-49.
- Dahwal V, Sharman AK, Deka D, et al. The obstetric outcome following treatment in a cohort of patients with antiphospholipid antibody syndrome in a tertiary care J Postgrad Med. 2011; 57: 16-19.
- Hidaka T, Suzuki Efficacy of filtration leukocytapheresis on rheumatoid arthritis with vasculitis. Ther Apher. 1997; 1: 212-214.
- Tuscano JM, Sands B cell reductive therapy with rituximab in the treatment of rheumatoid arthritis. Biologics. 2009; 3: 225-232.
- Malchesky PS, Smith JW, Koo A, et al. Uncontrolled trial of cryofiltration in rheumatoid J Clin Apher. 1988; 4: 156-165.
- Yu X, Mia J, Tian J, et al. A controlled study of double filtration plasmapheresis in the treatment of active rheumatoid J Clin Rheumatol. 2007; 13: 193-198.
- Onuma S, Yamji K, Kempe Investigation of clinical effect of large volume leukocytapheresis on methotrexate-resistant rheumatic arthritis. Ther Apher Dial. 2006; 10: 404-411.
- Caldwell J, Gendreau RM, Furst D, et al. A pilot study using staph. Protein A column (Prosorba) to treat refractory rheumatoid arthritis. J Rheumatol. 1999; 26: 1657-1662.
- Youssef P, Kennedy D. Arthritis in pregnancy: the role and safety of biological agents. Obstet Med. 2009; 2: 134-139.
- Hashizume M, Uchiyama Y, Horai N et al. Toclizumab, a humanized anti-interleukin-6 receptor antibody, improved anemia in monkey arthritis by suppressing IL-induced hepcidin production. Rheumatol Int. 2010; 30: 917-923.
- Bodaghi B, Grendron G, Wechsler B, et al. Efficacy of interferon alpha in the treatment of refractory and sight threatening uveitis; a retrospective monocentric study of 40 patients. Br Ophthalmol. 2007; 91: 335-339.
- Imrie FR, Dick D: Biologics in treatment of uveitis. Curr Opin Ophthalmol. 2007; 18:481-486.
- Malchesky PS. Therapeutic Apheresis: Why? Ther Apher Dial. 2015; 19: 417-426.