Therapeutic Apheresis, Immunosuppression, and Human Monoclonal Antibodies in Neurologic Diseases
Author'(s): Rolf Bambauer1*, Ralf Schiel2, Octavio J. Salgado3 and Richard Straube4
1 University of Saarland, formerly: Institute for Blood Purification, 66424 Homburg, G/Saar, Germany.
2 Clinic for Metabolic Diseases, Medigreif Inselklinik, 17424 Heringsdorf, Germany.
3 School of Medicine, Universidad Católica de Cuenca, Ecuador.
4 INUSpherese®, INUS Medical Center, Tagesklinikum, 93413 Cham, Germany.
*Correspondence:
Rolf Bambauer, MD, PhD, Frankenstrasse, 66424 Homburg/ Saar, Germany
Received: 21 Dec 2023; Accepted: 18 Jan 2024; Published: 25 Jan 2024
Citation: Rolf Bambauer, Ralf Schiel, Octavio J. Salgado, et al. Therapeutic Apheresis, Immunosuppression, and Human Monoclonal Antibodies in Neurologic Diseases. Clin Immunol Res. 2024; 8(1): 1-12.
Abstract
Therapeutic plasma exchange with hollow fiber modules is used since 45 years and in combination with immunosuppressive therapies and/or human monoclonal antibodies a steady increase in survival rates over the last decades. Therapeutic apheresis is accepted as supportive therapy in all severe neurological diseases such as in acute or chronic inflammatory demyelinating polyneuropathy, myasthenia gravis, multiple sclerosis, Refuse’s disease, Rasmussen encephalitis and others. Infection with COVID-19 can exacerbate and aggravate the neurological diseases due to autoimmune etiology. The therapy is the same like for the neurological diseases. Other therapy strategies are different human monoclonal antibodies with or without therapeutic apheresis. The knowledge of immunology and molecular biology of different neurological diseases are discussed in relation to the rationale for apheresis therapy and its place with other modern therapy strategies, and the pathogenetically aspects are demonstrated. Therapeutic apheresis has shown to effectively remove all autoantibodies and others toxins from blood and lead to rapid clinical improvement. The guidelines of the Apheresis Application Committee of the American Society for Apheresis are cited for neurological diseases, which could be treated with therapeutic apheresis.
Keywords
Abbreviations
AAC: American Applications Committee, AB: antibodies, AchR: acetylcholine receptor, ADEM: acute disseminated encephalomyopathy, AIDP: acute inflammatory demyelinating polyneuropathy, ASFA: American Society for Apheresis, BW: body weight, CIDP: chronic inflammatory demyelinating polyradiculoneuropathy, CNS: central nervous system, COVID-19: coronavirus disease, CSF: cerebrospinal fluid, GABHS: Group-A beta-hemolytic streptococcus, GBS: Guillain-Barre´ syndrome, FFP: fresh frozen plasma, EBV: Epstein Barr Virus, HGF: hepatocyte growth factor, HMA: human monoclonal antibodies, HP: hemoperfusion, IA: Immunosuppression, Il-1β: interleukin-1beta, IVIG: intravenous immunoglobulin, LA: lipoprotein apheresis,
LEMS: Lambert-Eaten myasthenic syndrome, LRP4: low-density lipoprotein-related protein, MFS: Miller-Fisher syndrome, MG: myasthenia gravis, MIP-1α, -1β: Macrophage inflammatory Protein- 1α-1β, MRI: magnetic resonance imaging, MS: multiple sclerosis, MuSK: muscle-specific kinase, NMJ: neuromuscular junction, PA: phytanic acid, PANDAS: Pediatric autoimmune neuropsychiatric disorders associated with Streptococcal infections, PiA: picolinic acid, PrA: pristanic acid, RE. Rasmussen encephalitis, RG: recommendation grade, SC: Sydenham´s chorea, Th3, Th2: T helper cell type1 and 2, TNF-α: tumor necrosis factor, TPE: therapeutic plasma exchange, TA: therapeutic apheresis.
Introduction
Up today therapeutic apheresis (TA) has been proved itself in a series of immunological, metabolic diseases, intoxications and others. With the introduction of hollow fiber modules in TA a complete separation of the corpuscular components from the plasma was shown and due to increased blood flow rate and higher efficacy. Therapeutic apheresis summarizes various extracorporeal blood purification techniques that removes inflammatory mediators, antibodies (ab) and other toxic substances, which are pathogenic in various diseases such autoimmune or non-immunological diseases [1]. Membrane separation techniques are simple and safe to apply and can be competitive to other plasma separation and treatment technologies [2]. With the adsorption technologies the most selective separation of plasma components are allowed without the use need of any substitution solution [3].
The advantages of membranes plasmapheresis its simplicity to use with blood pumps and no white blood cell or platelet loss, compare with centrifuges. Furthermore, cell damage, especially to thrombocyte, occurs less using membranes than centrifuge for cell separation. No advantage is that TA using centrifuges has a shorter treatment times such as TA using hollow fibers. Important is to keep the blood levels with antibodies and/or pathogenic substances on a very low level over long time during TA treatment. Than the substances that should be eliminated could invade into the intravascular space and could be eliminated by the membrane separators.
The therapeutic plasma exchange (TPE) equipment is not perfect, because the filtered plasma fractions have to be discarded, and substitution solutions supplemented with human albumin, plasma substitute, or fresh frozen plasma (FFP) are necessary to replace the discarded fractions. Therefore semi-selective, and selective adsorption methods are available without the need of a substitution solution.
The TPE has a variety of indications in neurology, nephrology, hematology, endocrinology, cardiology, pulmology, dermatology,oncology, infectiology and intoxications [4]. Only a few controlled trials are available that are of adequate statistical power to allow definitive conclusions to be reached regarding the therapeutic value of TA. The relative rarity of most of the disorders are under investigations. Many investigators have to composite understandably grouped heterogenous diseases together, often retrospectively, and used historical controls. In the most neurologic disorders besides TA an immunosuppression therapy and/or human monoclonal antibodies (HMA) are necessary. Therapeutic apheresis, which is indicated in neurologic disorders, includes TPE, immunoadsorption (IA) with organic or synthetic adsorbers, which contain staphylococcal protein A or synthetic peptide-goat-antimouse, which works like a mini-receptor together with epitope, and adsorbers with covalently tryptophan [3]. Further TA methods, which are indicated in neurologic disorders, are whole blood adsorption (hemoperfusion, HP), lipoprotein apheresis (LA), and others.
Neurological disorders constitute the largest group of indications for TA [5]. A poor prognosis, and a high mortality rate are combined with severe central nervous system (CNS) involvement. The first-lines of treatment in these diseases are high dose steroids and cyclophosphamide, oral or intravenous. Therapeutic apheresis, intravenous immune globulin (IVIG), thalidomide, intrathecal treatment, a HMA may be valuable in treatment resistant, and severe cases.
In view of the pathological aspects, the authors try to give an overview of immunologic and non-immunologic neurological diseases, in which TA, immunosuppression, and/or HMA are indicated. For those neurological disorders for which TA is indicated, the guidelines on the use of TA from the American Applications Committee (AAC) of the American Society for Apheresis (ASFA) are cited (Table 1) [6,7].
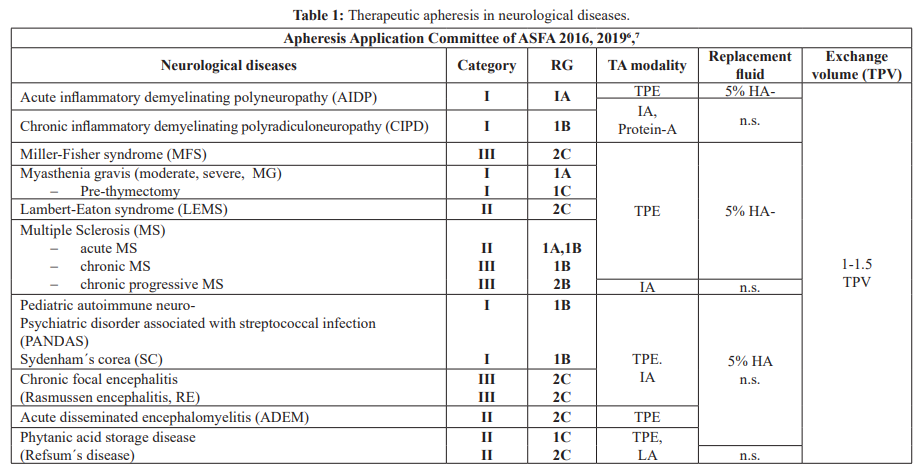
Category 1: accepted for TA as first line therapy; Category II: accepted for TA as second-line therapy; Category III: not accepted for TA, decision should be individualized; Category IV: not accepted for TA, IMB approval is desirable if TA is undertakenâ¶,â·.
TA: therapeutic apheresis, RG: recommendation grade, TPE: therapeutic plasma exchange, IA: immunoadsorption, LA: lipid apheresis, 5% HA: 5% human albumin electrolyte solution, n.s.: no substitution, TPV: total plasma volume.
Acute Inflammatory Demyelinating Polyneuropathy (AIDP),
Guillain-Barre´ Syndrome (GBS)
Acute inflammatory demyelinating polyneuropathy is an auto aggressive disorder that develops subsequent to infectious diseases and because of other noxae [8]. The AIDP is an acute polyradiculitis, which mostly affects the distal and proximal muscles of the extremities, and the trunk muscles and can progress with severe ascending paralysis, ending in respiratory paralysis [9]. An inflammatory, predominantly demyelinating polyneuropathy have the most patients. This disease is acute progressive, which leads to rising paralysis, and reaches its height in one to two weeks, and 25 % of all these patients require artificial ventilation. Regardless of gender or age every year in the industrial nations, AIDP occurs in one out of 50,000 persons [9]. In many patients, an antecedent infection by campylobacter jejuni leads to the production of antibodies directed against certain epitopes of the bacterium that also destroy the myelin sheath of the peripheral nerve, which is described as molecular mimicry [10]. However, the pathophysiology mechanism has not been established completely. The spectrum of organism responsible for infections can trigger AIDP ranges from Epstein-Barr virus to mycoplasma, herpes zoster, mumps virus borrelia, HIV to the corona virus [11- 13]. Acute inflammatory demyelinating polyneuropathy directly attacks the myelin sheath, resulting in segmental demyelination and remyelination.
Triggering causes for AIDP are: Antibodies against peripheral nerves, in particular against. In serum; other inflammatory mediators; a disorder in cell-related immunity [14]. Between the 2nd and 4th week of illness, a spontaneous recovery could be, and in 75% of patients, it can even occur after several months of illness. Lethality is between 5% and 25% after one year, due to remaining damage and relapses [8]. The differentiation between axonal and myelin lesions in end-stage acute polyneuropathy can be carried out with electro-diagnostic study. However, current electro- diagnostic criteria have some limitations in diagnosing axonal GBS [14]. The axonal type of GBS is pathophysiological characterized not only by axonal degeneration, however, also by reversible conduction failure. A correct diagnosis includes antiganglioside antibody and other ab tests [15,16]. There are also seronegative AIDP patients [17]. During the pandemic of coronavirus, there are few reports of AIDP associated with COVID-19 [13]. Especially after COVID-19 vaccination, in some patients were acute-onset chronic inflammatory demyelinating polyneuropathy observed. The patients were treated with oral prednisolone, azathioprine, IVIG and/or TA [18,19].
The rational for TA is based on humoral and cellular immune dysfunction in AIDP [20]. In combination or alone IVIG has also been shown to be effective in the treatment of ADIP. All three modalities of TPE, IVIG and conservative therapies were effective as treatment in AIDP, which was shown in large international randomized study [21]. The TPE was superior than IVIG, and the combination of both was better than either of the treatment alone [22,23]. A combination of TPE or IA following by IgG (0.4 g/kg BW for 5 days) may be superior to TPE alone [24]. It was possible to reduce the costs of the treatment of GBS with TPE by between 30 to 40% in America, due to the shorter periods of inpatient treatment and shorter duration of artificial respiration [8]. The AAC of the ASFA has given the AIDP the category I with the recommendation grade (RG) 1A (Table 1) [6,7].
In recent years, various human monoclonal antibodies were introduced successfully in the treatment of AIDP or refractory diseases in combination with immunosuppressive therapies [25,26]. The biologicals such as rituximab, brentuximab, or pembrolizumab and others in combination with immunosuppressives therapies were successfully in the treatment of AIDP [27,28]. However, further controlled and randomized studies are necessary to find out what therapy is superior.
Chronic Inflammatory Demyelinating Polyradiculoneuropathy (CIDP)
Chronic inflammatory demyelinating polyradiculoneuropathy is an uncommon progressive or relapsing paralyzing disease caused by inflammation of the peripheral nerves [6]. Neurologic symptoms are decreased sensation, diminished or absent reflexes, elevated cerebrospinal fluid level, and evidence of demyelination [7]. In CIDP, cellular and humoral components of the immune system attack myelin on large peripheral nerve fibers, leading to demyelination that manifests as weakness, numbers, paresthesia, and sensory ataxia [29]. An axonal loss occurs secondary to demyelination is a poor prognosis, if the disease progresses [29,30]. The CIDP is an acquired disease of the peripheral nervous system has probably an autoimmune pathogenesis. In most patients, the nature of the responsible auto-antigens is unclear. However, the frequency of such antibodies is significantly higher in CIDP than in normal control patients [31].
The CIDP is an inflammatory demyelination, which manifests as slowed conduction velocities, temporal dispersion, and conduction block nerve on nerve conduction studies and as segmental demyelination, onion-bulb formation, and endoneurial inflammatory infiltrates on nerve biopsies [32]. The clinical symptoms are weakness or sensory ataxia, etc. However, the clinical presentation and course variable extremely [1]. The prevalence of CIDP have ranged from 1.9 cases per 100.000 persons (Australia), to 3.6 cases per 100.000 persons (Italy) to 7.7 cases per 100.00 persons (Norway) to 8.9 per 100.000 persons (Rochester, Minnesota) [33]. More than 50% of the CIDP patients cannot walk unaided when symptoms are at their worst. All treatments which reduce the inflammation influence positively the CIDP, however, it is not clear which treatment is the best [34].
A short-term efficacy of IVIG, prednisone and TPE have confirmed in different clinical trials. In the absence of better evidence about long-term efficacy, corticosteroids or IVIG are usually cyclophosphamide, cyclosporine, other immunosuppressive agents, and interferon–β and α and HMAs such as rituximab, especially in treatment-resistant CIDP [29,30,35]. Besides rituximab, proteasome inhibitors such as bortezomib in combination with rituximab and cyclophosphamide could stabilize the majority of CIDP patients [36].
During the COVID-19 pandemic different acute and chronic demyelinating neuropathies after MIP-1α infection and exacerbations of neuroimmunology diseases, remains speculative. In larger case series, perhaps it could be clarified that SARS- CoV-2 infection might be a possible precipitating factor for clinical worsening in immune-mediated polyneuropathies [37- 39]. To summarize the therapy of the last years, IVIG and/or corticosteroids should be considered in sensory and motor CIDP, and IVIG should be the initial treatment in pure motor CIDP. If IVIG and corticosteroids are ineffective TA should be considered [31]. If the response is inadequate or maintenance doses of the initial treatment are high, combination therapy or adding an immunosuppressant or immunomodulatory drugs could be considered, and symptomatic treatment and multidisciplinary management should be considered. Especially in severe and treatment-resistant cases HMAs and/or proteasome inhibitors are indicated [38].
The AAC of the ASFA has given AIDP and CIDP the category I with 1A and 1B, respectively (Table 1) [6,7]. The main etiology of CIDP is autoimmune attack on the peripheral nerves. The humoral and cell-mediated immune response follow an increase of inflammatory cytokines, such as HGF, TNF-α, IL-1β, MIP-1α, and MIP1β [7]. Therapeutic apheresis removes these inflammatory cytokines quickly and the immune response normalize, and can accelerate motor recovery, decrease time on the ventilator, and speed attainment of other clinical symptoms [6]. The Cochrane Neuromuscular Disease group showed that TPE or IA in AIDP and CIDP are the most effective when initiated within the first 7 days of disease onset, and IA has been increasingly recognized alternatively to TPE for AIDP and CIDP [40]. However, larger controlled randomized studies are necessary to show the best and more effective therapies.
Miller-Fisher syndrome (MFS)
The MFS is characterized by acute onset of ophthalmoplegia areflexia. It is considered a variant form of GBS syndrome and has a close association with the presence of the anti-GQ1b antibody. Therefore, the efficacy of treatment with TPE and/or IVIG have to be proved. Some reports of the response of patients with MFS to TPE would be consistent with the pathogenic role for the anti- GQ1b antibody. There are some MFS patients without antibodies [41].
Miller-Fisher syndrome has a prevalence of one in 1,000,000 worldwide. The disease of MFS is higher in Asian than in Western populations [42]. Patients with MFS had deviated T-helper Typ-1 (Th3) / T-helper Type-2 (Th3) polarization and TA can shift Th3- dominant status to Th3-dominant status in patients with MFS [12]. Therapeutic apheresis can remove humoral factors including anti- GQ1b, and may induce a shift of the Th3/Th3 cytokine-producing cell balance in peripheral blood. During the pandemic, there are many case and small series reports of GBS and MFS in COVID-19 patients and by the clinical suggestion of treating neurological complications with IVIG [43,44]. Especially after COVID-19 vaccination, patients developed MFS who are treated with IVIG [45,46]. Miller Fisher syndrome is generally self-limited and has excellent prognosis [47]. In the guidelines of the AAC of the ASFA, the MFS has the category III and the RF 2C (Table 1) [6,7].
Myasthenia Gravis (MG)
Myasthenia gravis is caused by autoantibodies, which are directed against acetylcholine receptors of the skeletal muscles. The acetylcholine receptor antibodies (Ach-R-ab) belong to a heterogenous group of polyclonal abs. They are directed against various sections of the post-synaptic receptor molecule [12]. Normal nerve transmission from motor nerves to striated muscle is interrupted, due to blockage of the receptors. Myasthenia gravis primarily affects the muscles of the eyes, esophagus, and respiratory muscles, as well as the extremities.
Myasthenia gravis is an autoimmune disorder, and has the prevalence of 85-125 per million, and an annual incidence of 2-4 per million [48]. The disease is characterized by muscle weakness and fatigability. Patients with MG should be classified into subgroups to help with therapeutic decisions and prognosis. Subgroups are patients with muscle-specific kinase (MuSK) and the low-density lipoprotein-related protein (LRP4) antibodies [49,50]. The MuSK, a transmembrane tyrosine kinase, is expressed predominantly at the postsynaptic membrane of the neuromuscular junction (NMJ), and binds LRP4 and transmits an agrin-mediated signal for the clustering of AchR [51]. The LRP4 protein belongs to a family of proteins that has been recently identified as the receptor for the neural agrin that can activate MuSK [52].
Infection such as corona virus disease can exacerbate and aggravate neurological diseases due to autoimmune etiology like myasthenia gravis [53]. A COVID-19 infection can increase the risk of MG, respiratory failure, and mortality rate due to cytokine storm in MG patients. The therapies of COVID-19 patients with MG are based on the therapy recommendations for COVID-19 and the MG therapies.
The therapies are thymectomy and the application of cholinesterase-blocking substances [54]. In severe progression, immunosuppressives are also given to suppress. Especially in cases of severe, previously therapy-resistant progression, TA has been implemented with good results [55,56]. With TPE a rapid elimination of autoantibodies results, which follows in an improvement in clinical symptoms within hours to days. Immunosuppressive drugs target autoantibody production but can take months to have an effect. Therapeutic apheresis and IVIG have more rapid effect than immunosuppressive therapy [57]. The rationale for TA is to remove circulating antibodies. Therapeutic apheresis is the first-line therapy in acute attacks (Table 1). Seropositive and seronegative patients respond to TPE. Therapeutic plasma exchange is especially used in myasthenic crisis, perioperatively for thymectomy, or as a further therapy to other therapies to maintain optimal clinical status, and TPE works rapidly, clinical effect can be seen within 24 hours but may take a week [6]. The benefits of TPE will likely subside in 2 – 4 weeks, if immunosuppressive therapies are not initiated to keep antibody levels from reforming. Immunosuppressives in combination with TPE seems to be successful.
The new developed biologics offering powerful agents with favorable safety and tolerability profiles, which target key effector mechanisms in MG and could be long-standing clinical remission [58]. Rituximab, eculzumab, belimumab and ravulizumab, are used in clinical trials of patients with refractory MG and showed successful results [59-61]. These HMAs are used most in combination with the new complement inhibitors and neonatal Fc receptor blockers and immunosuppression [61,62]. However, further controlled, randomized studies are necessary.
Lambert-Eaten myasthenic syndrome (LEMS)
Lambert-Eaton myasthenic syndrome is very rare antibody- mediated autoimmune disease that is caused by serum autoantibodies and results in muscle weakness and autonomic dysfunction [63]. Like MG, LEMS is based on a disorder of the transmission of neuromuscular excitation in which cases no acetylcholine is released. By an autoimmune attack, LEMS is caused against presynaptic voltage gated calcium cannels and is characterized by late onset of fatigue, skeletal muscle weakness, weight loss, automatic dysfunction, and areflexia [64]. Lambert- Eaton syndrome presents itself in two forms: the paraneoplastic form, resulting mainly from small cell lung carcinoma, and the underlying autoimmune form [65].
The rationale is similar to that in MG, patients strength should be improved by the removal of the pathogenic antibody to voltage- gated calcium channel. Patients are treated long-term with a combination of corticosteroids and immunosuppressive therapy has failed TPE been attempted (Table 1) [66,67]. There are only case series, which have suggested some benefit by TPE. The treatment of the underlying cancer is besides the removal of the etiological agent is the most important therapy [65]. Rituximab binds CD20 antigen on B cells and has widely used in a variety of autoimmune disorders and various cancer diseases, and has been the subject of extensive application in MG and LEMS and appeared to be associated with improvement in patients with MG [67]. Other immune checkpoint inhibitors such as durvalumab or pembrolizumab were successfully applicated in LEMS [68,69]. However, further controlled studies must show the effectiveness in larger groups of patients.
Multiple Sclerosis (MS)
Multiple sclerosis is an autoimmune disorder affecting the central nervous system, and a relapsing chronic demyelinating disease and the most common cause of neurologic disability in young adults [70]. Worldwide, there are more than one million afflicted with MS. There are affected 120,000 to 140,000 patients with MS, alone in Germany, and in the United States, there are more than 300.000 patients. In children and adolescents, MS is also diagnosed. Estimates suggest that 8.000 to 10.000 children, up to 18 years, in the United States have MS, and another 10, 000 to 15,000have experienced at least one symptom suggestive of MS [12].
The pathophysiologic aspects of MS as an autoimmune disease is based on the following characteristics [22]:
- HLA association and genetic predisposition: T cell subject and cytokine correlation with disease activity,
- Clinical response to immunosuppression, immune activators, and biologics [71],
- Analogies with experimental autoimmune encephalitis,
- Cerebrospinal oligoclonal IgG bands,
- Central nervous system pathology using immunocytochemistry techniques,
- Evidence of intrathecal synthesis of tumor necrosis factor beta in MS, and the level of TNF-α in cerebro-spinal fluid mays correlate with the severity and progression of disease and reflect histologic disease activity in MS,
- Increased levels of gamma interferon correlate with the disease worsening [22].
The pathogenesis of MS, as autoimmune disease, is not clearly understood. Multiple sclerosis is epidemiologically a heterogenous disease influenced by genetic factors, such as the association with HLA-DRB!*15:01, and environmental factors, which include vitamin D level, obesity, smoking and Epstein Barr Virus (EBV) infection [70]. The prognosis of MS depends on the subtype and progression of the tissue [72]. Therapeutic plasma exchange may be benefit in MS patients by removing antibodies such as antimyelin antibody, or by modulating immune response. In the early MS, there have been four immunopathologic patterns [6]. The characteristics of demyelination for each pattern are: T cell/ macrophage-associated, antibody/complement-associated, distal oligodendrogliopathy, and oligodendrocyte degeneration. The B cells act as antigen-presenting cells to activate T cells and produce pro-inflammatory, such as interleukin-6, interferon-γ, and tumor necrosis factor, and anti-inflammatory cytokines (interleukin-10) that regulate the immune process. The source of mature plasma cells that secrete antibodies are these cells. B cells participate in the pathogenesis of the disease through multifunctional mechanism [73,74].
The rational for treatment of MS with TA is the presence of these circulating antimyelin antibodies, non-antibody demyelinating factors, aquaporin-4-specifix serum antibodies, and neuroelectric blocking factors [75]. These antibodies will be removed by TPE and other humoral factors from the circulation safely and effectivity. Therapeutic plasma exchange has shown to increase the number and percentage of suppressor T cells and decrease the helper cells in MS patients, too. The removing factors and toxic substances are following by an effectively decreasing the ratio of elevated helper/inducer to suppress/cytotoxic cell [76]. This is important, because the T cells play a pivotal role in the pathogenesis of MS [8]. Therapeutic apheresis and IA showed high efficacy and good tolerability [77]. Children with MS should be treated with corticosteroids. If this therapy do not bring enough improvement, other treatments such as IVIG, interferon β1a, and TPE are indicated to treat-to-treat MS attacks. Therapeutic plasma exchange is also been used in drug removal in MS with natalizumab which develop progressive multifocal leukoencephalopathy. It is a severe opportunistic brain infection caused by virus, which is a known complication of natalizumab therapy [6].
The AAC of the ASFA has given the acute attacks of MS the category II and the RG 1 A, 1B, the chronic MS and the chronic progressive MS the category III and the RG 1B, respectively 2B (Table 1) [6,7]. In the treatment are used TPE, IA, double filtration plasmapheresis which are safely and effective [77-81].
During the pandemic with COVID-19, SARS-CoV-2 infections in neurological diseases and after COVID-19 vaccination, a seroconversion were observed [82,83]. The anti-CD29 therapies in double vaccinated MS patients increase significantly post- infection as in the control group [84], Anti-CD20 therapies in double-vaccinated patients do not preclude an appropriate SARS-CoV-2 antibody response post-infection. Patients with MS exposed a wide spectrum of MS immunotherapies have important implications for treatment-specific COVID-19 clinical guidelines [83]. B cell depleting therapies are reassuring as at least partial protection form more severe COVID-19 outcome can be expected [85].
Rituximab showed efficacy in the treatment of MS. Other HMAs such as ocrelizumab, work by eliminating selected pathogenetic cell populations, and showed beneficial effects on relapsing MS and have partial effects on primary MS [86]. Monoclonal antibodies also carry the risk of infusion/injection-related reactions primarily in early phases of treatment [71]. Further anti-CD20 antibodies have been introduced in the treatment of MS, which are ofatumumab, or ublituximab, a new glycoengineered, chimeric anti-human CD20 [87]. Further studies here are necessary to find the benefit for patients with MS.
Pediatric Autoimmune Neuropsychiatric Disorders Associated with Streptococcal Infections (PANDAS); Sydenham´s chorea (SC)
Pediatric autoimmune neuropsychiatric diseases associated with streptococcal infections and Sydenham´s are post infectious neuropsychiatric diseases. These both diseases have neuropsychiatric symptoms, which typically follow Group-A beta hemolytic streptococcus (GABHS) infections. If this pathogenesis is postulated, Streptococcal antigens induce antineural antibodies by an abnormal immune response [6]. With childhood-onset neuropsychiatric GABHS infection has been associated. Acute and dramatic are the onset of PANDAS which present with emotional/ mood lability, attention deficit, deterioration of handwriting,separation anxiety, tactile/sensory defensiveness, enuresis,cognitive deficits, and motor hyperactivity [88].
In childhood SC is the main common acquired chorea. Chorea, hypotonia, and emotional lability are the major clinical manifestations. The duration of SC is several months with recurrence rate of about 20% [6]. For PANDAS and SC, the mean ages of onset are 6.8 and 8.4 years old, respectively. The diagnosis of SC is made exclusively by clinical presentations and a history of rheumatic fever. Choreatic movements are rapid, and affect the face, trunk, and the extremities. Group-A beta hemolytic streptococcus are associated with PANDAS, and is not associated with rheumatic fever. Elevated or increasing streptococcal antibody titers are shown in laboratory tests, but an elevated titer does not necessarily indicate a recent streptococcal infection. With at least two episodes of neuropsychiatric symptoms as well as negative throat culture or stable titers during times of remission, the presence of streptococcal infection in PANDAS is associated. Antibiotics have been used extensively in the treatment for PANDAS and cognitive behavioral therapy. Severe forms of SC are treated with diazepam, valproic acid, carbamazepine, or haloperidol [6]. If all these therapies fail, corticoids may be introduced, and other therapies are IVIG, TPE, tonsillectomy, cognitive behavior therapy [89]. The efficacy of penicillin prophylaxis in preventing symptoms exacerbations in children with PANDAS remains doubtful, while children with SC require long-term penicillin prophylaxis to reduce the risk of rheumatic carditis. Intravenous IgG (1 g/kg/day for 2 days) or TPE has been shown to reduce symptom severity or shorten the course in severe symptomatic or refractory patients with PANDAS or SC. After the conservative therapy has been exhausted, or the first-line therapy in situations of life threatening functional impairment, TPE is indicated in severe cases extreme cases [90]. The treatment of TPE is daily or every other day for five or six procedures over 7 to 14 days. The AAC of the ASFA has given Pandas or SC the category I with RG 1B (Table 1) [6,7]. However, TA should be reserved for treatment of children and adolescents who are severely affected by PANDAS. It appears to be safe well-tolerated, and beneficial treatment option in these patients. Besides the first-line therapy with steroids, IVIG, and TA, the second-line treatments are cyclophosphamide or rituximab [91]. Newer experimental concepts for neurological disorders are gene therapies [92].
Chronic focal encephalitis (Rasmussen encephalitis, RE)
The Rasmussen disease is a chronic focal encephalitis, and characterized by intractable focal seizures and slowly progressive neurological deterioration [6]. In childhood is typically the onset, mean age 6.8 ± 5.1 years, however a similar syndrome has been described in adults, too. The etiology of this disease is unknown, but antecedent infection with Epstein-Barr virus, herpes simplex, enterovirus, or cytomegalovirus has been implicated. In three adult patients with Rasmussen´s encephalitis, cytomegalovirus genome has been found in resected cortical tissue. The cerebrospinal fluid analysis in most patients is normal. Mild lymphocytic pleocytosis and elevated protein may be found. In Rasmussen encephalitis is the important symptom epilepsy uncontrollable with anticonvulsant drugs, progressive hemiparesis, and progressive unilateral cerebral atrophy. In the affected cerebral hemisphere, there is a progressive loss of function [6]. Rasmussen disease is a cell-mediated immune attack on one cerebral hemisphere, through the inciting antigen remains unknown [93].
To control the disease or to stop its progression, anticonvulsants are necessary but not always effective. Steroids, tacrolimus, IVIG, and TA were applied in Rasmussen disease [94]. However, subtotal, functional complete hemispherectomy can markedly reduce seizure activity in a majority of patients. This results in permanent contralateral hemiplegia corticosteroids and IVIG given for up two years in tapering schedule to diminish epilepsia and other symptoms [6,95].
Antibodies in Rasmussen encephalitis against neural molecules, and autoantibodies can be produced in the CNS after cytotoxic T cell-mediated neuronal damage [8]. The Rasmussen encephalitis has received the category III with the RG 2C from the AAC of the ASFA (Table 1). The rationale for TA is as follows. Neuropsychological assessment may be helpful in evaluating patients with slowly progressive disease to determine whether TPE is effective in postponing surgical therapy. An initial course of TPE may be followed by 2 days of IVIG 1 g/kg/day. Monthly IA of 1.5-2 TPV per treatment has been effective in some patients [6]. In patients with RE and GluR3 antibodies, TA may support a benefit. The frequency is every other day. After initial 5-6 TPE treatments over 10-12 days, subsequent courses of TPE (with or without IVIG) may be performed at 2-3 month intervals as empirically needed. Immunosuppressive drugs may increase the interval between courses. Besides steroids, IVIG, tacrolimus, TPE, and HMAs, such as rituximab in the treatment of Rasmussen disease, were introduced. Especially with rituximab some patients has a significant benefit [96]. However, until to date, there is no definitive consensus on treatment, proposed strategies ranging from acute or chronic immunotherapy to hemispherectomy [97].
Acute disseminated encephalomyopathy (ADEM)
Acute disseminated encephalomyopathy is an acute inflammatory monophasic demyelinating disease that effects the brain and spinal cord, which typically occurs after a febrile, often presumed to be viral prodrome or vaccination [6]. In absence of specific biologic markers, the diagnosis of ADEM must be done with clinical and radiologic features [98]. Ataxia, weakness, dysarthria, and dysphagia accompanied by change in mental status are typically presentation for the multifocal neurological deficits. It is most a monophasic illness that lasts from 2 to 4 weeks. Most affected are children and young adults. The differentiation of ADEM from the first attack of multiple sclerosis has prognostic and therapeutic implications. The ADEM has these features that help to distinguish it from MS, which are florid polysymptomatic presentation, lack of oligoclonal band in cerebrospinal fluid (CSF), predominance of MRI lesions in the subcortical region with relative sparing of the periventricular area, and complete or partial resolution of magnetic resonance imaging (MRI) lesions during convalescence [6,8].
The ADEM is an acute, rapidly progressive autoimmune disorder in which either microbes or immunization have an important role to play [99]. During the pandemic, ADEM has also been associated with COVID-19 infection and rarely with COVID-19 vaccination [99]. During prolonged hospitalizations neurological complications of COVID-19 may develop, which are particularly difficult to evaluate and appreciate in the critically disease [100]. Cas series have shown the causal association between ADEM and COVID-19 vaccination [101].
The first-line therapy in ADEM are corticosteroids, which has recovery and result in clinical improvement in up to 60 % of patients. In patients who do not respond to corticosteroids IVIGs are indicated [6,102]. In neurologic diseases that are presumed to be immunologically mediated, TPE is used and has a clearly defined role. Therapeutic apheresis removes presumed offending antibodies as well as through immunomodulation. The category II for TPE with the RG 2C after the AAC of the ASFA is assigned on paucity of data (Table1) [6,7]. Typically is TPE given in steroid refractory severe cases, or in life-threatening cases TPE should be considered early in the disease course [103,104]. In children with severe ADEM, TPE appears to be of benefit if introduced as early as possible the frequency is every other day between 3 to 6 treatments [105]. Human monoclonal antibodies seem to be of benefit only in ADEM with autoimmune origin.
Phytanic Acid Storage Disease (Refsum´s Disease)
Refsum´s disease, heredopathia atactica polyneuritiformis, is a rare recessive autosomal inherited metabolic disorder, based on an isolated lack of the enzyme, which results in phytanic acid (PA) being stored in the body and causing corresponding symptoms [106]. Retinitis pigmentosa, anosmia, deafness, chronic sensory- motor neuropathy, ataxia and the accumulation of PA in blood and human tissues are the clinical symptoms [107,108]. A significant improvement in Refsum´s disease can be achieved by removal of PA through TPE and a PA-reduced diet [109]. The first and important therapy step in Refsum´s disease is dietary restriction. The average daily intake of PA is 50-100 mg/day, and should be reduced to 10-20 mg/day. Phytanic acid is exclusively of exogenous origin and levels of PA >800 μmol/L is not uncommon. Poorly metabolized PA, pristanic acid (PrA), and picolinic acid (PiA) accumulate in fatty issues, myelitis sheaths, heart, kidney and retina, leading to retinitis pigmentosa, peripheral dissociative polyneuropathy, cerebral ataxia, “sailors walk”, renal, cardiac and liver impairment 65% of plasma PA and PrA are localized within VLDL, LDL, HDL lipoprotein particles [110]. The dietary restriction of PA is mostly not sufficient to prevent acute attack and stabilize the progressive course [109,110]. If PA is reduced to below 500 mg/L by TPE, clinical improvement is achieved. The experience with black cumin oil, nigella sativa in a dose of 3 g/day shows a support and regression of some malnutrition effects in PA restricted dietary and a supportive effect to membrane differential filtration [110].
The AAC of the ASFA has given the Refsum´s disease the category II with the RG 1C-2C (Table 1) [6,7]. Therapeutic plasma exchange can reduce the elevated plasma levels of PA, and can avoid acute attacks or exacerbation of the disease as well as for maintenance therapy. In humans the normal plasma PA level is <33 μmol/L, in Refsum´s disease, symptomatic levels range from 700-800 μmol/L. Phytanic acid is also bound to plasma lipoproteins and triglycerides therefore LA and membrane differential filtration has been successfully used to treat Refsum´s disease patients [6,110].
The treatment of Refsum´s disease with TA vary. A normal course consists of 1-2 TPE per week for several weeks to months [6]. In some patients maintenance TPE continue with decreasing frequency over subsequent weeks to months. The monitoring of the patient´s PA blood level, clinical signs and symptoms and the need to control or prevent exacerbation of the disease determine the therapeutic strategy [110]. No cure exists for Refsum´s disease, however, phytanate blood levels in patients can be reduced by different methods of TA and a strict diet [111,112].
Other Neurologic Diseases
In the Stiff-Man syndrome were found autoantibodies to GABAergic neurons, and they could be removed by TPE, the patient improved [113]. For the treatment of the coagulopathy extensive blood and plasma exchange have been successfully implemented in children with meningococcemia [114]. Immunoadsorption or lymphocytapheresis have been applied in ataxic neuropathy and idiopathic hypertrophic cranial pachymeningitis, Fabry disease, acute transverse myelitis and subacute sclerotic panencephalitis with success [115,116]. Other neurological diseases, such as cryoglobulinemia, polyneuropathy, central nervous system systemic lupus erythematosus, acquired neuromyotonia, polymyositis/ dermatomyositis, polyneuropathy in paraproteinemia, neuropathy by hyperlipidemia, and encephalopathy in metabolic/hematologic diseases such as thyrotoxicosis, hepatic coma, and M. Moskowitz are diseases that involve more organ systems and are mentioned elsewhere. Therapeutic apheresis could be regarded as a support therapy to the current treatment strategies in the above mentioned neurological disease.
Closing Remarks
Only a few prospective controlled studies of TA in various diseases are available to allow definitive conclusions. The prognosis of immunological diseases with their varying organ manifestation has improved considerably in recent years due to very aggressive treatment schemes including TA, immunosuppressive drugs and biologic agents. As first-line therapy, TA, corticosteroids, IVIG, immunosuppressive drugs, HMAs, has been established in many neurological diseases. All TA methods are safe and effective procedures. Immune-mediated neurological diseases, which without these treatment strategies can lead to significant disability and in limited number of patients to death, has a better prognosis [117]. Several factors are dictated for a specific therapy in an individual patient, including patient´s comorbidity and the practice environment. New opportunities for target interventions could provide from an understanding of antibody responses and genetic backgrounds in immune-mediated neurological diseases.
With new developed disease-specific adsorbers and new hollow fiber modules as second membranes more and more immunologic and non-immunologic diseases can be treated with TA. Besides physical problems and technical ones such as the apparatus required, the vascular access plays an important role [118]. If TA is indicated it must be started early as possible in the treatment with an adequate exchange volume and a lowest possible extracorporeal volume [119].
For pediatric patients guidelines have been written for implementation [118,120]. For adults and pediatric patients, an adequate blood flow is required which can be achieved via a large- bore catheter in the internal jugular or subclavian vein, or via peripheral large veins [1]. Sterile procedures must be adhered to prevent catheter infection and sepsis, if a large-bore catheter is used [121]. A well-trained and experienced team can overcome technical problems in order to complete the procedure without complications. The most frequently observed adverse effects are vascular relative access insufficiency (2%), and mild hypotension (2%) [12,122]. Therapeutic apheresis methods can be safely delivered by the medical staff in hemodialysis departments. Therapeutic apheresis is indicated in severe cases if the immunosuppression therapy has failed. The AAC of the ASFA has given the category I to AIDP, CIPD, PANDAS, SC. The category II has received Lambert-Eaton myasthenic syndrome, acute MS, Refsum´s disease, and ADEM. The category III has the MFS, chronic and chronic progressive MS, and the Rasmussen encephalitis (Table 1) [6,7].
Human monoclonal antibodies such as rituximab, eculizumab, belimumab, and others showed clinical improvement, especially in severe and refractory immune-mediated neurological disorders [63]. More controlled multicenter studies are necessary. All mentioned TA methods are still complicated and very expensive. The costs must be reduced, this is a valid demand in view of the scare resources available in the healthcare systems. For all mentioned diseases the quotient relevant for cost-effectiveness assessment (cost of treatment – cost saved): (improvement in life quality) must discuss and calculated exactly by all involved persons. Every effort should be made to delay the progression of acute or chronic diseases. Therapeutic apheresis is clearly an important tool treatment of many complex conditions now and in future [123].
References
- Afzali M, Oveisgharan S, Rajabkhah S, et al. Complications of therapeutic plasma exchange in patients with neurological disorders. Curr J Neurol. 2020; 19: 8-12.
- Malchesky PS. Membrane processes for plasma separation and plasma fractionation Guideline Principles for clinical Ther Apher. 2001; 5: 270-282.
- Bambauer R, Schiel R, Lehmann B, et al. Therapeutic Apheresis Technical Overview. ARPN J Sci Technol. 2012; 2: 399-421.
- Balsa R. Therapeutic Plasma exchange An Indispensable Therapy for Severe Neurological J Crit Care Med. 2020: 6: 89-90.
- Shelat Practical considerations for planning a therapeutic apheresis procedure. Am J Med. 2010; 123: 777-784.
- Schwartz J, Padmanabhan A, Aqui N, et Guidelines on the use of Therapeutic Apheresis in Clinical Practice- Evidence-Based Approach from the Writing Committee of the Ameri9can Society for Apheresis. The Seventh Special Issue. J Clin Apher. 2016; 31: 149-162.
- Padmanabham A, Connelly Smith L, Aqui N, et Guidelines on the use of Therapeutic Apheresis in Clinical Practice- Evidence Based Approach from the Writing Committee of the American Society for Apheresis. J Clin Apher. 2019; M34: 171-354.
- Bambauer R, Latza R, Burgard D, et Therapeutic Apheresis in Immunological Renal and Neurological Disease. Ther Apher Dial. 2017; 21: 6-21.
- Ariga T, Yu RK. Antiglycolipid antibodies in Guillain-Barre syndrome and related diseases. Review of clinical features and antibody specifities. J Neurosci Res. 2005; 80: 1-17
- Miller AC, Rashid RM, Sinert Guillain-Barre Syndrome. Medicine specialities Emergency Medicine. Neurol update Apr. 2010.
- Mancardi GI, del Sette M, Primavera A, et al. Borrelia burgdorferi in fectipon and Guillain-Barre´ Lancet. 1989; 21: 985-986.
- Bambauer R, Schiel Therapeutic Apheresis in Neurology. J Clin Res Rep. 2020;
- Gagarkin DA, Dombrowski KE, Thakar KB, et al. Acute inflammatory demyelinating polyneuropathy or Guillain- Barre´ syndrome associated with Covid-19 a case J Med. 2021; 15. https://link.springer.com/article/10.1186/ s13256-02831-4.
- Koo YS, Shin HY, Kim JK, et Electro-diagnostic Features of Upper Extremity Sensor. 2016; 12: 495-501.
- Junichi K. Anti-Neurofscin 155 Antibody-Positive Chronic Inflammatory Demyelinating Polyneuropathy/Combined Central and peripheral Demyelination: Strategies for Diagnosis and Treatment Based on the Disease mechanism. Front Neurol. 2021; 12. https://doi.org10.3389/fneur.2021.665136.
- Kim S, Lee EK, Sohn E. Reversible conduction failure in acute inflammatory demyelinating polyneuropathy. Nature. 2022; 12: https://www.nature.com/articles/s41598-022-195447-0.
- Davis AJ, Fehmi J, Senec M. Immunoadsorption and Plasma Exchange in Seropositive and Seronegative Immune-Mediated J Clin Med. 2020; 9.
- Fotiadou A, Tsiptsios D, Karatzetzou S, et al. Acute-onset chronic inflammatory demyelinating polyneuropathy complicating SARS-CoV-2 infection and AD26.COV2.S vaccination report of two cases. Egyp J Neurol Psyciat Neurosurg. 2022; 58: 116. https://link.springer.com/ article/10.1186/s41983-022-00515-4.
- Kim S, Lee EK, Sohn E. Two case reports of chronic inflammatory demyelinating polyneuropathy after COVID-19 vaccination. J Kor Med Sci. 2023; 38: e57.
- Weinstein R. Is there a scientific rationale for therapeutic plasma exchange or intravenous immune globulin in the treatment of acute Guillain-Barre´ syndrome? J Clin Apher. 1995; 10: 150-157.
- Korintenberg R, Schessl J, Kirchner J, et al. Intravenous administered immunoglobulin in the treatment of childhood Guillain-Barre´- syndrome. A randomized trial. Pediatrics. 2005; 116: 8-14.
- Khatri BO. Therapeutic apheresis in neurologic disorders. Ther Apher. 1999; 3: 161-171.
- Dada MA, Kaplan Plasmapheresis treatment in Guillain- Barre´- syndrome. Potential benefit over IVIG in patients with axonal involvement. Ther Apher Dial. 2004; 8: 409-412.
- Haupt WF, Birkmann C, van der Ven Apheresis and selective adsorption plus immunoglobulin treatment in Guillain-Barre´ syndrome. Ther Apher. 2000; 4: 198-200.
- Vendramin C, Scully M. Indications of plasma exchange in combination with intravenous immunoglobulins or therapeutic monoclonal La Presse Medicale. 2019; 48: 354-359.
- Pelletier JPR, Mukhtar F. Chapüter 16 passive monoclonal and polyclonal antibodies therapies. Immunol Conc Transf Med. 2020; 251-348.
- Hashimoto R, Ueda T, Tsuji Y, et Successful treatment of Guillain-Barre´ syndrome inflammatory demyelinating polyneuropathy of pembrolizumab with a combination of immunoglobulins a case report. Clin Neurol. 2020; 60. https:// doi.org/10.5692/clinicalneurol.cn-001444.
- Sharma S, Panda S, Tiwari Dlemmas in acute-onset chronic inflammatory demyelinating polyneuropathy A new axonal variant. A Indian Acad Neurol. 2022; 25: 515-517.
- Dyck PJB, Iracy JA. History diagnosis and management of chronic inflammatory demyelinating polyneuropathy. Mayo Clin Proc. 2018; 93: 777-793.
- Ryan M, Ryan SJ. Chronic Inflammatory Demyelinating Polyneuropathy Considerations for Diagnosis Management and Population AM J Manag Care. 2018; 24: S371-S379.
- Hughes Management of chronic inflammatory demyeli- nating polyradiculoneuropathy. Drugs. 2003; 63: 275-287.
- Dyck PJB, Tracy JA. History Diagnosis and Management of Chronic Inflammatory Demyelinating Polyradiculoneuropathy. Mayo Clin Proc. 2018; 93: 777-793.
- Said Chronic inflammatory demyelinating polyneuropathy.Neuromusc Disord. 2006; 16: 293-303.
- Oaklander AL, Lunn MPT, Hughes RAC, et Treatments for chronic inflammatory demyelinating polyradiculoneuropathy CIDP an Overview of systematic reviews. Crtochane Datab Syst Rev. 2017. https://doi.org/10.1002/14651858.CD010369. pubs2.
- Jiao L, Xiang Y, Li S, et al. Efficacy of low dose rituximab in treatment-resistant CIDP with antibodies against NF-155. J Neuroimmunol. 2020; 345: https://doi.org/10.1016/j. neuroim.2020.577280.
- Pitarokoili K, Yoon MS, Kröger I, et al. Severe refractory CIDP a case series of 10 patients treated with bortezomib. J Neurol. 2017; 264: 2010-2020.
- Taga A, Lauria G. COVID-19 and the peripheral nervous system. A 2-year review from the pandemic to the vaccine J Peripher Nerv Syst. 2022; 27: 4-30. https://doi.org/10.1111/ jms.12482.
- Erin D, Hartmann E, Cortes-Penfield N, et al. Acute and Chronic Demyelinating Neuropathies After COVID-19 Vaccination: A report of 4 J Clin Neuromusc Dis. 2023; 24: 147-156.
- Abu-Rumeileh S, Garibashvili T, Ruf W, et al. Exacerbation of chronic inflammatory demyelinating polyneuropathy in concomitance with with COVID-19: J 2020; 418: 117106.
- Oji S, Nomura Immunoadsorption in neurological disorders. Trans Spher Sci. 2017; 56: 671-676.
- Winer Bicjerstaff´s encephalitis and the Miller-Fisher syndrome. J Neurol Neurosurg Psychiat. 2001; 71: 433-435.
- Ooi ST, Ahmad A, Yaakub A. Recurrent Miller Fisher Syndrome. Cureus. 2022; 14: e261192.
- Mangoti P, Pesavento V, Buoite A, et al. Miller Fisher syndrome diagnosis and treatment in a patient with SARS- CoV-2. J 2020. https://doi.org/10.1007/s133656- 020-008589.
- Zhao H, Shen D, Zhou H, et al. Guillain-Barré syndrome associated with SARS-CoV-2. Lancet 2020; 19: 383-384.
- Nantsue K, Takahashi M, Itaya S, et A case of Miller Fisher syndrome with delayed onset peripheral facial nerve palsy after COVID-19 vaccination a case report. BMC Neurol. 2022; 22: 309.
- Yamakawa M, Nakahara K, Nakanishi T, et Miller Fisher Syndrome Following against SARS-CoV-2. Int Med. 2022; 61: 1067-1069. https://doi.org/10.2169/ internmalmedicine.8851-21.
- Noioso CM, Bevilacqua L, Acerra GM, et al. Miller Fisher syndrome an update narrative Front neurol. 2023; 14: 1250774.
- Romi F, Gilhaus NE, Aarli JA. Myasthenia gravis clinical immunological and therapeutic Acta neurol Scand. 2005; 111: 134-141.
- Gihaus NE, Verschuuren JJ. Myasthenia gravis subgroup classification and therapeutic Lancet neurol. 2015; 14: 1023-1036.
- Behin A, Le Panse New Pathways and Therapeutic Targets in Autoimmune Myasthenia gravis. J Neurolmusc Dis. 2018; 5: 265-277.
- Lin W, Burgess RW, Dominguez B, et Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature 2001; 410: 1057-1064.
- Zanhg B, Tzartos JS, Belmiezi M, et al. Autoantibodies to lipoprotein-related protein 4 in patients with double-seronegative myasthenia Arch Neurol. 2012; 69: 445-451.
- Tugasworo D, Kurnanto A, Retnaningsih a, et al. The relationship between myasthenia gravis and COVID-19a systematic Egypt J Neurol Psychiat Neurosurg. 2022; 58: 83.
- Schneider-Gold C, Toyka Myasthenia gravis Pathogenesis and Immunotherapy. Dtsch Ärztebl. 2007; 104: a420-426.
- Bachmann K, Burkhardt D, Schreitert J, et al. Thymectomy is more effective than conservative treatment for myasthenia gravis regarding outcome and clinical Surgery. 2009; 392-396.
- Fernandez-Fournier M, Kerguelen A, Rodriguez de Rivera FJ, et Therapeutic plasma exchange for myasthenia gravis, Guillain-Barré syndrome, and other immune-mediated neurological diseases, over a 40-year experience. Exp Rev Neurotherap. 2022; 22: 897-9903. https://doi.org/10.1080/14737175.2022.214827.
- Mantegazza R, Bonanno S, Camera G, et al. Current and emerging therapies for the treatment of myasthenia gravis. Neuropsychiat Dis Treat. 2022; 7: 151-160. https://doi. org/10.2147/NDT.S8915.
- Dalakas MC. Advances in the therapeutic algorithm for myasthenia gravis. Nat Rev Neurol. 2023; 19: 393-394.
- Howard Myasthenia gravis the role of complement at the neuromuscular junction. Ann NY Acad Sci. 2017; 1412: 113-128.
- Mantegazza R, Antozzi When myasthenia gravis is deemed refractory clinical signposts and treatment strategy. Adv Neurol Dis. 2018. https://doi.porg/10.1177/1756285617749134.
- Saccà F, Pane C, Espinosa PE, et al. Efficacy of innovative therapies in myasthenia gravis a systematic review meta- analysis and network meta-analysis. Eur J Neurol. 2023. https://doi.org/10.1111/ene.15872.
- Uzawa A, Utsugisawa Biological therapies for myasthenia gravis. Exp Opin Biolog Ther. 2023; 23: 253-260. https://doi. org/10.1080/14712598.2023.2184257.
- Vershuuren JGM, Wirtz PW, Titulaer MJ, et al. Available treatment options for the management of Lambert-Eaton myasthenic syndrome. EXP Opin Pharmacother. 2006; 7: 1323-1336.
- Huang L, Luo YB, Yang Autoimmune Channelopathies at Neuromuscular Junction. Front Neurol. 2019; 10: 516.
- De Souza FF, Trevisani JP, Ibiapina dos Reis F. Clinical Pathophysiological and Electrodiagnostic Aspects of Lambert- Eaton Myasthenic Syndrome. Topics in Autonomic Nervous System. Chapter Metrics Overview. 2023.
- Motomura M, Hamasaki S, Nakane S, et al. Apheresis treatment in Lambert-Eaton myasthenic syndrome. Ther Apher. 2004; 4: 287-290.
- Pascuzzi RM, Bodkin CL. Myasthenia Gravis and Lambert- Eaton Myasthenic Syndrome New Developments in Diagnosis and Neuropsychiat Dis Treat. 2022; 18: 3001-
- https://doi.org/10.2147/NDT.S296714.
- Machiyama H, Minami S. Durvalumab for Extensive-Stage of Small-Cell Lung Cancer With lambert-Eaton Myasthenic Syndrome. J Med Cases. 2023; 14: 71-75.
- Lee JH, Baek SK, Han JJ, et al. Lambert-Eaton myasthenic syndrome LEMS in a patient with lung cancer under treatment with pembrolizumab a case study. J Chemother. 2023; 35: 275-280. https://doi.org/10.1080/1120009X.2022.2073162.
- Yang JH, Rempe J, Whitmire N, et al. Therapeutic Advances in Multiple Sclerosis. Front Neurol. 13: 2022. https://doi. org/10.3389/fneur.82496.
- Kranjc N, Bsteh G, Berger T, et al. Monoclonal Antibodies in the Treatment of Relapsing Multiple Sclerosis an Overview with Emphasis on Pregnancy Vaccination and Risk Management. Neurotherapeutics. 2022; 19: 753-773.
- Fymat Multiple Sclerosis III Treatment and prognosis. J Neurol Psychol Res. 2023; 4.
- Shen P, Filatreus Antibody-independent functions of B cells a focus on cytokines. Nat Rev Immunol. 2015; 15: 441-451.
- Li RT, Rezk A, Healy Cytokine-defined B cell responses as therapeutic targents in multiple sclerosis. Front Immunol. 2016; 6: 1-10.
- Khatri BO. Therapeutic apheresis in multiple sclerosis and other central nervous system disorders. Ther 2000; 4: 263-270.
- Magna SM, Pittcock SJ, Lennon VA, et NMO-IgG status in fulminant inflammatory CNS demyelinating disorders. Arch Neurol. 2009; 66: 964-966.
- Dorst J, Fangerau M, Tarnu D, et Safety and efficacy of immunoadsorption versus plasma exchange in steroid- refractory relapse of multiple sclerosis and clinically isolated syndrome A randomized parallel-group controlled trial. Lancet. 2019; 16: 98-106.
- Straube R, Müller G, Voit-Bak K, et Metabolic and Non-Metabolic peripheral Neuropathy Is there a Place for Therapeutic Apheresis. Horm Meta Res. 2019; 51: 779-784.
- Lin Y, Oji S, Miyamoto K, et al. Real-world application of plasmapheresis for neurological disease Results from the Japan-Plasmapheresis Outcome and Practice Patterns Study. Ther Apher Dial. 2022; 27: 123-135.
- Bunganic R, Blahutova S, Revendova K, et al. Therapeutic plasma exchange in multiple sclerosis patients with aggressive relapse an observational analysis in a high-volume center. Nature. 2022; 18374.
- Kaleta FP, Jain S, Uqdah H, et Myasthenia Gravis requiring Therapeutic Plasma Exchange less than one month after coronary stent placement: A Pharmacologic dilemma. J Am Coll Cardiol. 2023; 81: 3815.
- Wu X, Wang L, Shen L, et al. Response of COVID-19 vaccination in multiple patients following disease-modifying therapies A meta-analysis. eBioMedicine, 2022.
- Sabatino JJ, Mittl K, Rowies WM, et Multiple sclerosis therapies differentially affect SARS-CoV-2 vaccine-induced antibody and T cell immunity and function. JCI Insight. 2022; 7: e156978.
- Habek M, Zeljko C, Mlakar AS, et al. Humoral and cellular immunity convalescent and vaccinated COVID-19 people with multiple sclerosis Effect of disease modifying Mult Scler Relat Dis. 2022; 59: 103682. https://doi.org/10.1016/j. msard.2022.103682.
- Stoll S, Desai S, Levit E. A retrospective evaluation of seroconversion after COVID-19 during the early Omicron wave in fully vaccinated multiple sclerosis patients receiving anti-CD20 therapy. Mult Scler Relat Dis. 2023; 104574. https://doi.org/10.1016(j.msard.2023.104574.
- Mulero P, Midaglia L, Montalben X. Ocrelizumab a new milestone in multiple sclerosis Ther Adv Neurol Dis. 2018. https://doi.org/10.1177/1756286418773025.
- Giedratiene N, Kaubrys G, Kizlaitiene, et al. Therapeutic plasma exchange in Multiple Sclerosis in Patients with Abolished Interferon-beta Bioavailability. Med Sci Monit. 2025; 21: 1512-1519.
- Bambauer R, Schiel R. Therapeutic Apheresis in Pediatrics with Neurological and Hematological Diseases. J Clin Exp Immunol. 2020; 5: 156-172.
- Sigra S, Hesselmark E, Bejerot Treatment of PANDAS and PNS a systematic review. Neurosci Biobehavior Rev. 2018; 86: 51-65.
- Latimer ME, L´Etoile N, Seidlitz J, et Therapeutic Plasma Apheresis as Treatment for 35 Severely III Children and Adolescents with Pediatric Autoimmune Neuropsychiatric Disorders Associated with Streptococcal Infections. J Child Adoles Psychopharmac. 2015; 25: 70-72.
- Bien CG, Bien Autoimmune encephalitis in children and adolescents. Neurol Res Pract. 2020; 2. https://doi. org/10.1186/s42466-019-0047-8.
- Shaqhcheraghi SH, Ayatollahi J, Lofti M, et Gene therapy for neuropsychiatric disorders potential targets and tools. CNS Neurol Dis. 2023; 22: 51-65.
- Cay-Martinez K, Hickman RA, McKhan GM, et Rasmussen encephalitis An update. Semin Neurol. 2020; 40: 201-210.
- Chamlagain R, Shah S, Youssef P, et Different modalities of the treatment of Rasmussen encephalitis A systematic review of case reports of a rare disease. Browse. 2022. https://orcid. org/0000-0002-8203-3329.
- Varadkar S, Bien CG, Kruse CA, et al. Rasmussen´s encephalitis clinical features pathobiology and The Lancet Neurol. 2014; 13: 195-205.
- Jagtap SA, Patil S, Joshi A, et al. Rituximab in Rasmussen´s encephalitis A single center experience and review of the literature. Epilep Behav Rep. 2022; 19: 100540. https://doi. org/10.1016/j.ebr.2022.100540.
- Castellano JF, Meyer FA, Lado A Case Serious of Adult- onset Rasmussen´s Encephalitis Diagnostic and Therapeutic Challenges. Front Neurol. 2017. https://doi.org/10.3389/ fneur.2017,00564.
- Tenembaum S, Chitnis T, Ness J, et al. Acute disseminated encephalitis. Neurology. 2007; 68.
- Permezel F, Borojevic B, Lau S, et al. Acute disseminated encephalomyelitis ADEM following recent Oxford/ AstraZenenca COVID-19 Foren Sci med Pathol. 2021; 18: 74-79.
- Ross Reichard R, Kashani KB, Boire NA, et Neuropathology of COVID-19 a spectrum of vascular and acute disseminated encephalomyelitis ADEM-like pathology. Acta Neuropathol. 2020; 140: 1-6.
- Nabizadeh F, Noori M, Rahmani S, et Acute disseminated encephalomyelitis ADEM following COVID-19 vaccination A systemic review. J Clin Neurosci. 2023; 111: 57-70. https:// doi.org/10.1016/jocn.2023.03.008.
- Parsons T, Banks S, Bae C, et COVID-19-associated acute disseminated encephalomyelitis ADEM. J Neurol. 2020; 267: 2799-2802.
- Pohl D, Tenembaum S. Treatment of acute disseminated encephalomyelitis. Cur Treat Opt 2012; 14: 264-275.
- Fjellbirkeland CW, Szpirt WM, Borresen M. The role of plasmapheresis in severe acute disseminated encephalomyelitis with clinical findings of transverse Ther Apher Dial. 2023. https://doi.org/10.1111/1744-9987.14059.
- Moussa T, Baker L, Chehab A, et Plasma Exchange for Autoimmune Encephalitis and Demyelinating Encephalomyelitis. Diagnostic and Therapeutic Challenges. J Neuroinfect Dis. 2017; 8: 262-269.
- Frankovich J, Swedo SE, Murphy TK, et al. Clinical Management of Pediatric Acute-onset Neuropsychiatric Syndrome Part II Use of Immunomodulatory Therapies. J Child Adolesc Psychopharma. 2017; 27: 1-16.
- Choksi V, Hoeffner E, Karaaslan E, et Infatile Refsum Disease Case Report. A J Neuroradiol. 2003; 24: 2082-2084.
- Hagermann RJ, Hall DA, Coffey S, et al. Treatment pod fragile x-associated tremor ataxia syndrome. Clin Interv Aging. 2008; 3: 251-262.
- Verny C, Prundean A, Nicolas G, et Refsum´s disease may mimic familial Guillain-Barre´ syndrome. Neuromuscuc Dis. 2006; 16: 805-808.
- Straube R, Gäckler D, Thiele A, et al. Membrane differential filtration is safe and effective for the long-term treatment of Refsum syndrome an update of treatment modalities and pathophysiological cognition. Transfus Apher Sci. 2003; 29: 85-91.
- Zolotov D, Wagner S, Kalb K, et al. Long-term strategies for the treatment of Refsum´s disease using therapeutic J Clin Apher. 2012; 27: 99-105.
- Wegrzyn AB, Herzog K, Gerding A, et Fibroblast-specific genome modeling an imbalance in amine acid metabolism in Refsum disease. FFBS J. 2020; 287. https://doi.org/10.1111/ febs.15292.
- Brashear HR, Philipps LH. Autoantibodies to GABergic neurons an response to plasmapheresis in stiff-man syndrome. 1991; 41: 158. https://doi.org/10.1212/ WNL.41.10.1588.
- Churchwell KB, Mc Manus ML, Kent P, et al. Intensive blood and plasma exchange for treatment of coagulopathy in meningococcemia. J Clin Apher. 1995; 10: 171-177.
- Sato T, Nakaqo T, Baba M, et al. Immunoadsorption plasmapheresis in acute ataxic Ther Apher. 1998; 2: 71-73.
- Yammamoto T, Geigo K, Suzuki A, et Long- term improvement of idiopathic hypertrophic cranial pachymeningitis by lymphocytapheresis. Ther Apher. 2000; 4: 313-316.
- Beydoun SR, Brannagan TH, Donfrio P, et al. Chronic inflammatory Demyelinating Polyradiculoneuropathy 101-Pitfall and Pearls of Diagnosis and Treatment. US Neurology. 2017; 13: 18-25.
- Shelat Practical considerations for planning a therapeutic apheresis procedure. Am J Med. 2010; 123: 777-784.
- Bambauer R, Schiel R, Salgado OJ, et al. Therapeutic Apheresis, Immunosuppression and Human Monoclonal Antibodies in Clin Immunol Res. 2023; 7: 1-16.
- Galacki DM. An Overview of therapeutic apheresis in pediatrics. J Clin Apher. 1997; 12: 1-3.
- Bambauer R, Schiel R, Salgado OJ. Large-bore catheters as vascular access for extracorporeal detoxification methods: Advantages, disadvantages and necessary improvements. HSOA J Angiol Vasc Surg. 2022; 1000090.
- Wright E, Dillon MJ, Tullus K. Childhood vasculitis and plasma exchange. Eur J Pediatr. 2007; 166: 145-151.
- Malchesky Therapeutic Apheresis Why. Ther Apher Dial. 2015; 19: 417-426.